Murray (Nobel Prize 1992) first tried azathioprine in humans and added corticosteroids to the regimen (14). The improved results obtained by combination of azathioprine with corticosteroids ushered in the era of clinical renal transplantation in the early 1960s, through the successful studies of Thomas Starzl (18) and Murray et al. (14). Innovative therapies, such as antithymocyte globulin (ATG or ALG) (1970s), cyclosporine and anti-CD3 monoclonal antibody (OKT3) (1980s), mycophenolate, tacrolimus (1990s), and others, have markedly increased success. Recent clinical trials with protocols to induce mixed or complete chimerism have shown promise of achieving Medawar’s goal of specific tolerance without immunosuppression (19,20,21).
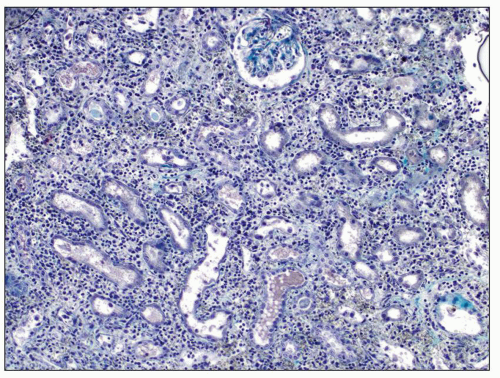 FIGURE 29.1 Photomicrograph of the first renal transplant biopsy (27). A living related kidney transplant (mother to son) was done under no immunosuppression in Paris in 1952 at the Necker Hospital under the supervision of Jean Hamburger. The biopsy was done for anuria on day 21 and shows a diffuse infiltrate of activated mononuclear cells, capillaritis with intracapillary neutrophils, glomerulitis, interstitial hemorrhage, and severe tubular injury, typical of combined acute TCMR (T-cell mediated rejection) and AMR. Trichrome-stained slide. (Courtesy of Christophe Legendre, Paris.) |
patients with delayed graft function (DGF). For treatment of acute rejection episodes, the usual first defense is a short course (2 to 3 days) of high-dose steroids orally (prednisolone) or intravenously (methylprednisolone), followed if necessary by rescue with ATG. Additional FDA-approved drugs include rapamycin (sirolimus, a blocker of IL-2 signaling and cell proliferation) and monoclonal antibodies to the IL-2 receptor (basiliximab [Simulect]), CD52 (daclizumab, CAMPATH1) and CD20 (rituximab [Rituxan]), and inhibitors of costimulatory signals (belatacept). All of these drugs have the potential for complications related to immunosuppression, and some cause nephrotoxicity, especially CNI.
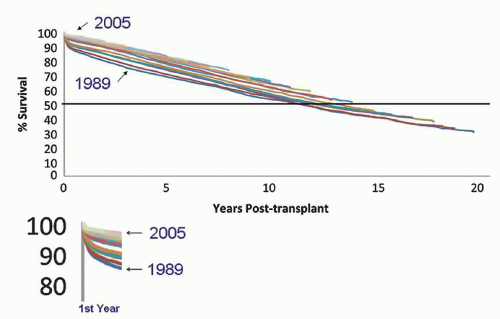 FIGURE 29.2 Kaplan-Meier plot of kidney graft failure by year of transplantation from 1989 to 2005 for 140,900 standard criteria deceased donor kidneys, censored for death due to other causes (38). Each colored line is a single year cohort from transplant year 1989 on the bottom to 2005 on the top. The half-life is indicated by the horizontal line at 50%. Most of the improved survival during this time period occurred in the first year posttransplant (shown in the insert). The slopes after 1 year are close to parallel, indicating little or no change in the rate of late graft loss. (Reprinted with permission from Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: A critical reappraisal. Am J Transplant 2011;11(3):450-62; copyright 2011, John Wiley and Sons.) |
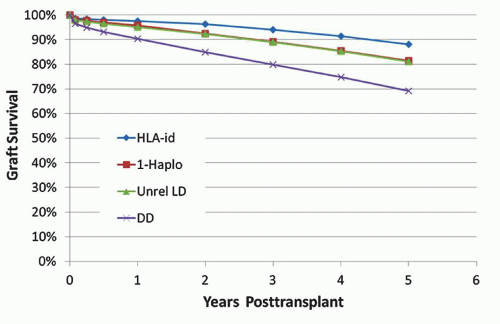 FIGURE 29.3 Graft survival according to the HLA match and type of donor. Survival of HLA-identical sibling grafts (HLA-id) is better than for related grafts with one HLA haplotype match (1-Haplo), demonstrating the importance of complete HLA matching. However, living unrelated grafts (Unrel LD) have better survival than deceased donor grafts (DD), indicating the state of the graft itself is also important. Living unrelated grafts have similar survival as one HLA haplotype-matched living grafts. Figure provided by Michael Cecka (University of California at Los Angeles) based on data from the Renal Transplant Registry of the Organ Procurement and Transplantation Network and United Network of Organ Sharing as of March 2011. N, Number of patients and T½, calculated half-life in years. |
importance of the MHC as a target in the immune response in chronic rejection.
of “n” biopsy cores can be calculated as 1-(1-sensitivity of a single core)n. Accordingly, if one core has a sensitivity of 90%, two cores have a predicted sensitivity of 99%, substantiating the conventional wisdom that recommends two cores.
the addition of AMR in 2003 (82). Banff scores the individual elements of the biopsy by light microscopy and uses these to classify rejection (Table 29.3). Combinations of individual scores are then used to define various categories of acute and chronic rejection. While many details are still being refined, Banff has had a beneficial effect in the standardization of definitions for publications and provides a stimulus for consensus development and translational research.
TABLE 29.2 Pathologic classification of renal allograft diseases | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Procurement biopsies are primarily collected to give information on the organ suitability for transplantation, and implantation biopsies provide insight into preexisting diseases relevant for comparative analyses posttransplantation; thus, the diagnostic implications of both biopsy types are similar. Since procurement biopsies are often evaluated on a rush basis by general surgical pathologists during off-hours in the frozen section laboratory and recorded under the donor’s name, the analysis is commonly rudimentary, and the results are often unavailable for subsequent graft management under the “new” recipient’s name. In some cases, both procurement and zero-hour implantation biopsies are collected. In general, guidelines for the interpretation and recording of donor biopsies have not been definitively established. Often, Banff scoring criteria are used, which can, unfortunately, easily lead to subsequent confusion during the evaluation of diagnostic graft biopsies since the scoring results of “old preexisting donor lesions” versus “new de novo posttransplant changes” are not distinguished from each other using ci, ct, and cv scores.
TABLE 29.3 Banff scores of individual features | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
mistaken for fibrosis or edema. Glomerular cellularity cannot be reliably assessed, although thrombi and crescents can be identified. The minimum number of glomeruli needed to correlate with outcome was found to be 25, and the minimum number required to obtain consistent results from paired biopsies was 15 (92). In our opinion, at least 25 glomeruli should be studied, from as deep in the cortex as is feasible. If a scar is sampled, as indicated by clusters of globally sclerotic glomeruli, this area should be noted, but it should be treated separately in the analysis to avoid overstating the percent sclerosis. Most importantly, a wedge biopsy is not representative, since it includes mostly outer cortex, the zone where glomerulosclerosis and fibrosis due to vascular disease is most severe. Intimal fibrosis, in contrast, most prominently affects arcuate and larger-caliber arteries and therefore is underrepresented in a wedge biopsy (93,94,95). If needle core biopsy samples are obtained on isolated/procured kidneys by the surgeons in the operating room with a so-called biopsy gun, then the tissue cores may show predominately renal medulla thereby limiting the diagnostic yield considerably. This problem can be avoided by “shooting” tangentially rather than perpendicularly into the “naked procured” organ. Good results were also reported using skin punch biopsy tools (94). In general, procurement biopsies are examined with H&E stains only; serial step sections and special stains (including trichrome incubation for the evaluation of sclerosis) are usually not performed, thereby further limiting the diagnostic yield.
vessels constitutes a contraindication to renal transplantation, organs with focal fibrin thrombi limited to the microvasculature can have an excellent prognosis, although these grafts may experience an initial period of DGF. Glomerular thrombi are found in 3% to 7% of donor biopsies, particularly in those originating from patients with head trauma (97,112,113). Thrombi in less than 50% of glomeruli after reperfusion had no effect on graft outcome in one series (114), but in another series of nine donor kidneys with glomerular thrombi (of unspecified extent), three had primary nonfunction (PNF) (97). Thrombi disappeared in five recipients with subsequent biopsies as soon as 8 days postgrafting, and long-term outcome was unaffected even in cases with “severe” microthrombosis (113). It is likely that unaltered fibrinolysis in the recipient results in full restoration of blood flow postgrafting. TMAs, in contrast, affecting small arteries with intramural vascular injury, extravasation of fragmented red blood cells, intimal remodeling, and swelling are poor prognostic indicators and should preclude transplantation. Anecdotal reports and personal experience suggest that eclamptic kidneys can fully recover (112). It is not established whether an occasional cholesterol embolus is a contraindication. However, donor-derived atheroembolization is often multifocal and associated with a high graft failure rate (115,116).
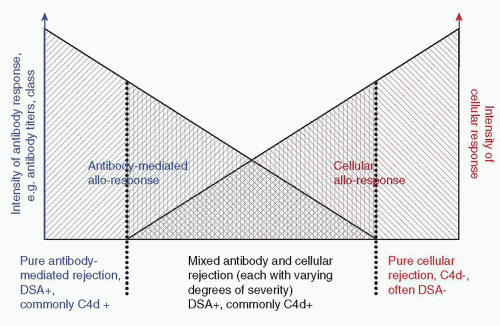 FIGURE 29.4 Schema of acute/active rejection. Acute rejection episodes can either be pure antibody- or pure cell-mediated events or also represent mixed rejection with varying degrees of antibody and cellular components. |
leukocytes into a mildly edematous interstitium and into the tubules (so-called tubulointerstitial cellular rejection; Banff category 4, type I rejection). In some cases, plasma cells dominate, that is, the so-called plasma cell-rich cellular rejection. The infiltrate can in more severe TCMR episodes also (or occasionally solely) affect arteries (so-called transplant endarteritis; Banff category 4, type II rejection) and glomeruli (so-called transplant glomerulitis; not specifically categorized in the Banff classification scheme). Inflammation is accompanied by signs of injury of the target cells, such as endothelial swelling, tubular cell activation, or apoptosis. Acute cellular rejection is often patchy and typically affects the cortex. Rejection-induced inflammation in the medulla is only seen in pronounced cases as a “spillover effect.” In general, medullary inflammation might have many etiologies such as PVN, pyelonephritis, allergic interstitial nephritis, etc., and it is least specific for rejection-induced injury. Diffuse and pronounced interstitial edema or hemorrhage is only seen in most severe forms of acute TCMR involving arteries and glomeruli. Such cases often also show concurrent AMR and C4d positivity.
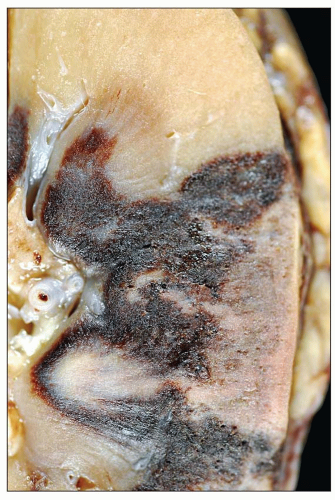 FIGURE 29.5 This transplant failed within few weeks postgrafting due to severe rejection with fibrinoid arterial wall necrosis (v3 lesion) and presumed mixed acute antibody- and cell-mediated rejection. Segments of hemorrhagic infarction in the cortex and medulla border on zones with normal-appearing renal parenchyma. Large arterial cross sections show some degree of arteriosclerosis but no gross evidence of thrombus formation. |
TABLE 29.4 Banff types of acute T-cell-mediated rejection | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
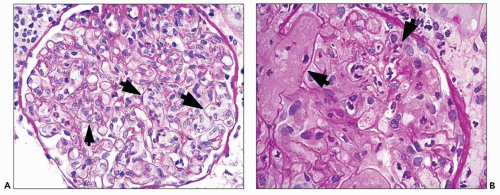 FIGURE 29.6 Transplant glomerulitis. A: Acute cellular rejection with transplant glomerulitis 6 days posttransplantation. Peripheral glomerular capillaries are dilated and “occluded” by mononuclear cell elements and swollen endothelial cells (arrows). The biopsy also showed transplant endarteritis and tubulointerstitial cellular rejection. This case of acute TCMR was not associated with acute AMR. The acute TCMR episode responded fully to thymoglobulin therapy. B: In comparison, a case of acute AMR, C4d-positive with transplant glomerulitis. This case shows mainly intracapillary polymorphonuclear leukocytes and fibrin microthrombi (compare to figure A); this case did not show concurrent acute CR. (PAS, ×500.) |
mesangiolysis. Intracapillary polymorphonuclear leukocytes and small fibrin thrombi can occasionally be seen; these latter features are more prominent in C4d-positive cases with (concurrent) AMR (Fig. 29.6B). Fibrinoid tuft necrosis, crescent formation, or GBM duplications are not features of rejection-induced glomerulitis. Glomerular lesions are best appreciated in 2-µm PAS-stained sections or silver stains with a good nuclear counterstain (GBM duplication characterizes transplant glomerulopathy and chronic rejection that can be associated with transplant glomerulitis in chronic active rejection episodes). Transplant glomerulitis with endothelial cells as a target of the cellular immune response occurs in approximately 60% of cases combined with other acute rejection-induced vascular lesions, that is, transplant endarteritis (53,157,158,159,163,164,165,166). In one series of 12 patients, 92% had associated endarteritis (165). In exceptionally rare cases, glomeruli can be solely affected without rejection-induced changes in other renal compartments (157,161,163). We found that approximately 20% of biopsies with transplant glomerulitis are “pure” Banff category 4 cellular rejection episodes lacking concurrent acute AMR/C4d positivity that is detected in the remaining cases. The predominance of intraglomerular monocytes (glomerular monocyte/T-cell ratio greater than 1) is more typical of acute AMR-induced transplant glomerulitis (167,168). In nonhuman primates protracted transplant glomerulitis evolved into chronic rejection/transplant glomerulopathy within 6 weeks (164).
cortex; proximal tubules are often spared and collecting ducts in the medulla hardly ever involved (175,176,177). Although plasma cells found in the so-called plasma cell-rich cellular rejection can be abundant in the interstitium (subtype of Banff category 4, type I rejection), they rarely invade tubules, and “plasma cell” tubulitis is uncommon. Occasionally scattered neutrophils can be seen in tubular lumens as a sign of acute injury. Small interstitial granulomas form adjacent to ruptured tubules and leakage of Tamm-Horsfall protein (uromodulin) into the interstitium (Fig. 29.10); this feature is nondiagnostic and can occur in many forms of tubular injury. Tubulitis in atrophic tubules, that is, less than 50% of the original diameter and markedly thickened TBM, is currently considered to be a nondiagnostic sign of parenchymal scarring; at present, this feature is not used to establish a diagnosis of acute TCMR (see Fig. 29.9C). However, this view may change in the future since there is increasing evidence that all tubulitis (in atrophic and nonatrophic tubules) and all interstitial inflammation (in scarred and nonscarred regions) is a sign of TCMR (see Fig. 29.9D) (149,178,179). The newly introduced Banff scoring entity of “total inflammatory (ti) score” may help assess the significance of tubulointerstitial inflammatory cell infiltrates (139). In some cases of acute TCMR, marked reactive atypia of tubular epithelial cells including multinucleation is seen. Such regenerative, ischemia-induced changes often occur in the setting of protracted severe rejection with marked edema, transplant endarteritis, and/or concurrent acute AMR/C4d positivity. Very pronounced tubular epithelial atypia is usually inconsistent with a diagnosis of “pure” tubulointerstitial cellular rejection, Banff category 4 type 1.
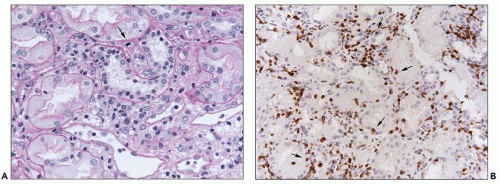 FIGURE 29.7 Several mononuclear cells are present in glomerular capillaries. This is below the threshold of transplant glomerulitis. (PAS stain, 400× original magnification.) |
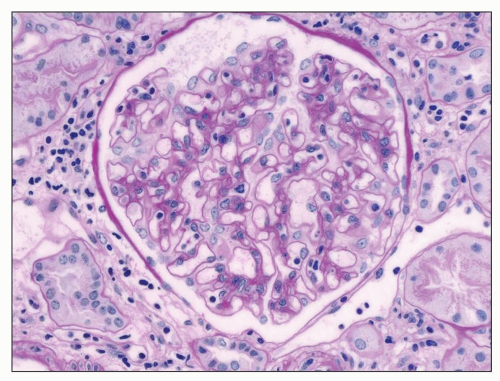 FIGURE 29.8 Acute tubulointerstitial cellular rejection with tubulitis. A: Activated mononuclear cells and edema are present in the interstitium. A mitotic figure is present in the infiltrate (arrow). (×40 PAS) B: A CD3 stain in another case shows abundant T cells in the interstitial and intratubular compartments/foci of tubulitis (arrows). (PAS stain, 40× original magnification. Immunoperoxidase ×200.) |
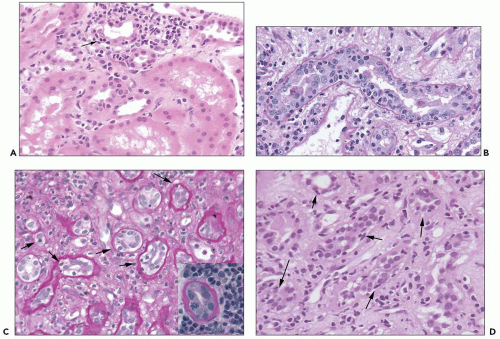 FIGURE 29.9 Diagnostic and nondiagnostic tubulitis. A: Diagnostic tubulitis: Over 10 mononuclear cells are in one tubular cross section (arrow). Surrounding tubules have zero to two infiltrating cells. (H&E ×400.) B: Diagnostic tubulitis: extreme tubulitis (t3) in a longitudinal section. (PAS ×400.) C: Nondiagnostic tubulitis: Inflammation in severely atrophic tubules is currently considered to be a nondiagnostic observation. Mononuclear cells (arrows) are in small tubules with thickened basement membranes. (PAS ×400.) The presence of occasional mitotic figures in “atrophic” tubules belies their designation of atrophy. (Insert, original magnification ×600.) D: Likely diagnostic tubulitis: Tubulitis is present in these smaller tubules with simplified, clear cytoplasm. However, the tubular diameters do not seem to be markedly reduced, and the TBM is not severely thickened. This focus of tubulitis possibly represents active rejection. (H&E ×400.) |
prominent and may indicate the presence of transplant endarteritis (Fig. 29.11). Inflammation in the setting of tubulointerstitial rejection (Banff category 4, type 1) is associated with relatively mild and often focal edema; marked diffuse edema usually indicates endothelial injury seen in cases with transplant endarteritis (Banff category 4, type II rejection) and/or concurrent acute AMR with C4d positivity. In acute TCMR, edematous regions often show dilatation of PTC and so-called PT capillaritis with intracapillary mononuclear cell elements (181). Polymorphonuclear PT capillaritis is rare and usually indicates acute AMR.
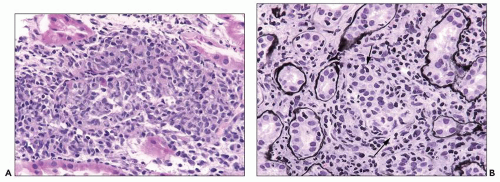 FIGURE 29.10 Severe tubulitis. A: Acute cellular rejection with tubular rupture and granuloma formation. (H&E ×400.) B: Partial dissolution and rupture of the tubular basement membrane can be appreciated in periodic acid-silver stains (arrows). (×400.) |
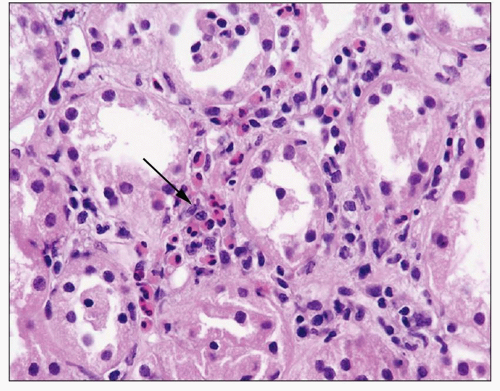 FIGURE 29.11 Acute cellular rejection with abundant eosinophils. Eosinophils (arrow) are about 20% of the infiltrate in this field. (H&E ×400.) |
of acute TCMR (Fig 29.13). Many terms have been used for this process, including endothelialitis, endotheliitis, endovasculitis, intimal arteritis, infiltrative and proliferative transplant vasculopathy, or endarteritis. We prefer endarteritis, because it emphasizes the type of vessel involved and the site of inflammation; also, more than the intima can be affected in some cases. The biologic and diagnostic significance of endarteritis was probably first noted and illustrated by Dammin in 1960 (28). The importance of this lesion has been emphasized for many years (202), and it is widely accepted as a feature of acute TCMR, particularly if transplant endarteritis is accompanied by tubulointerstitial cellular rejection (61,81,196). However, a considerable proportion of acute TCMR with transplant endarteritis also has concurrent acute AMR (142,150,151,153). On occasion, endarteritis can occur as an isolated event without tubulointerstitial changes. Since the endothelial cell layer is a key immunologic target in transplant endarteritis, the close association between endarteritis and glomerulitis is not surprising, and the detection of glomerulitis should always raise suspicion for transplant endarteritis.
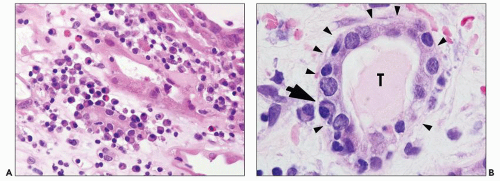 FIGURE 29.12 Plasma cell-rich acute TCMR. A: Acute tubulointerstitial cellular rejection rich in plasma cells (Banff category 4, type 1; C4d-negative, no DSA, no transplant endarteritis, no polyomavirus nephropathy) 8 years posttransplantation. Note abundant plasma cell aggregates in the edematous interstitium accompanied by scattered eosinophilic leukocytes. Plasma cells are “hugging” tubules, but they do not invade, that is, they do not cause “plasma cell” tubulitis. (H&E, ×640 oil.) B: This case of acute TCMR (C4d-negative, no DSA, no polyomavirus nephropathy) rich in plasma cells shows plasma cell tubulitis with one plasma cell (arrow) located inside the basement membrane (arrowheads) between tubular epithelial cells (T, tubular cross section). Plasma cell tubulitis is relatively uncommon. (H&E ×400.) |
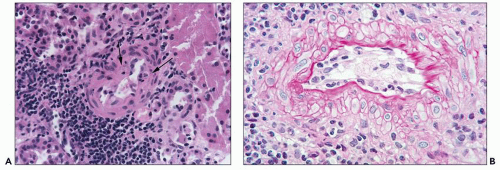 FIGURE 29.13 A, B: Acute cellular rejection with transplant endarteritis (Banff category 4, type 2 rejection). A: Small artery with few subendothelial mononuclear cells (arrows). (H&E ×400.) B: Small artery with many subendothelial mononuclear cells. The endothelial layer is not clearly defined and is probably partially denuded. Note: no thrombus formation and no inflammation in the medial smooth muscle layer. (PAS ×400.) |
becomes a major problem if endarteritis occurs as an isolated rejection event.
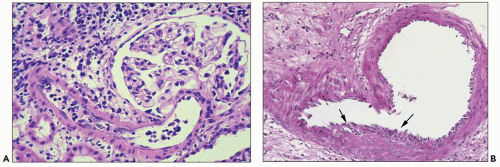 FIGURE 29.14 Acute cellular rejection with transplant endarteritis (Banff category 4, type 2 rejection). A: An afferent arteriole has a prominent mononuclear inflammatory cell infiltrate. This finding in a small-caliber artery is of similar significance as endarteritis affecting larger arteries; compare with B. (H&E ×400.) B: Arcuate size artery with endarteritis (arrows). (PAS ×200.) |
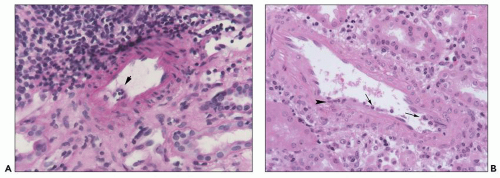 FIGURE 29.15 Acute cellular rejection with transplant endarteritis (Banff category 4, type 2 rejection). A: Minimum endarteritis. A single subendothelial infiltrating inflammatory cell is evident; endothelial cells are activated but intact. (PAS ×400.) B: Mononuclear cells mainly stick to the endothelium (arrows) and are only focally underneath the endothelium (arrowhead). Only subendothelial mononuclear inflammatory cells are diagnostic for transplant endarteritis. (H&E ×300.) |
transplant endarteritis, inflammation is typically limited to the intima/subendothelial zone sparing the medial smooth muscle layer. Transmural inflammation involving all layers of arterial walls including segmental fibrinoid necrosis can occur in severe cases of acute TCMR (Banff category 4 type 3 rejection) (Fig. 29.19); however, this feature is more often seen in biopsies with (concurrent) acute AMR and C4d positivity (150). Arteries located in or adjacent to the renal capsule can also be affected by endarteritis; however, diagnostic interpretation is challenging due to the altered blood flow in intracapsular vessels of allografts.
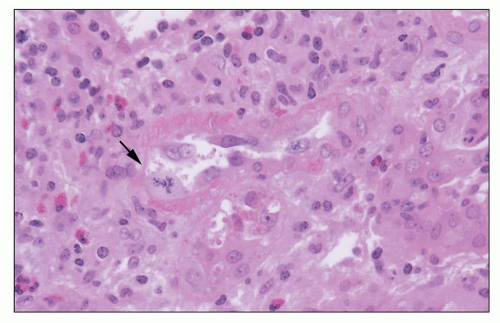 FIGURE 29.16 Acute cellular rejection with transplant endarteritis (Banff category 4, type 2 rejection). Small artery with reactive, enlarged, basophilic endothelial cell cytoplasm. An endothelial mitosis is present (arrow). Eosinophils are seen in the surrounding mixed mononuclear interstitial infiltrate. (H&E ×600.) |
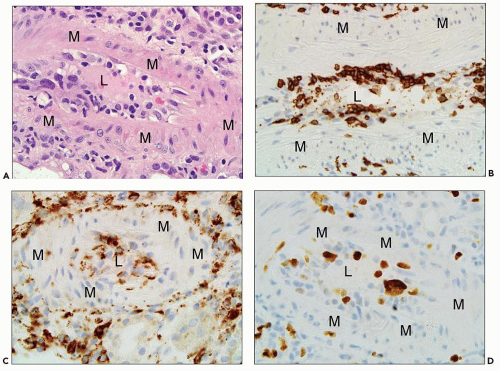 FIGURE 29.17 Acute cellular rejection with transplant endarteritis (Banff category 4, type 2 rejection). Seven days posttransplantation, this patient experienced diffuse tubulointerstitial cellular rejection, marked diffuse transplant glomerulitis, and marked transplant endarteritis (Banff type IIB rejection, C4d-negative). The patient was not sensitized, crossmatch and PRA negative with no DSA at the time of biopsy. Inflammation along the intimal layer of an interlobular artery (A) is rich in CD3 (B) and, to a lesser degree, also CD68 (C)-expressing cells that are mitotically active (Ki-67/MIB-1 positivity, D). |
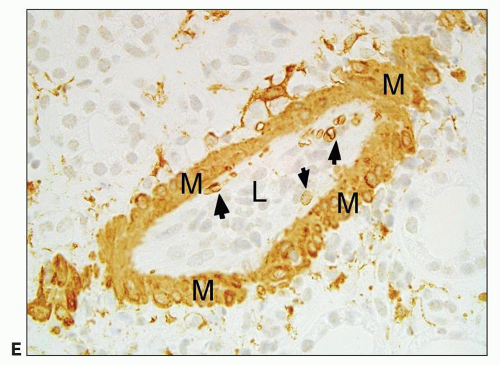 FIGURE 29.17 (Continued) Note: Even in cases of early transplant endarteritis, intimal inflammation contains scattered myofibroblasts expressing smooth muscle actin (SMA, arrows, E). Myofibroblasts can synthesize collagens and promote chronic vascular rejection in protracted/suboptimally treated rejection episodes. This patient responded fully to thymoglobulin therapy. ((A) H&E, (B-D) IHC on formalin-fixed and paraffin-embedded tissue, (B) anti-CD3, (C) anti-CD68, (D) Ki-67/MIB-1, (E) anti-alpha smooth muscle actin, ×600 oil. L, arterial lumen; M, medial smooth muscle layer.) |
are the dominant cell type in PT capillaritis suggesting in our experience concurrent acute AMR.
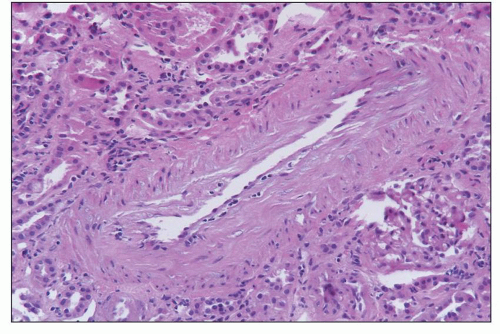 FIGURE 29.18 Acute cellular rejection with transplant endarteritis (Banff category 4, type 2 rejection). Subendothelial mononuclear cells are present in an artery (= acute rejection) superimposed upon preexisting arteriosclerosis (donor disease). This pattern can be confused with chronic rejection; elastic tissue stains usually help making a distinction (strong staining in cases of hypertension-induced arteriosclerosis vs. chronic vascular rejection largely lacking elastic tissue accumulations). (H&E ×200.) |
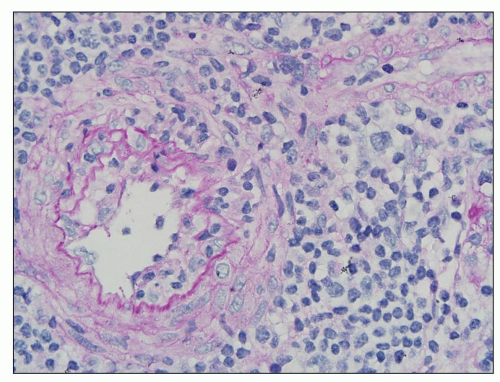 FIGURE 29.19 Acute cellular rejection with severe transplant endarteritis (Banff category 4, type 3 rejection). An interlobular artery has segmental transmural inflammation including destruction of the lamina elastica interna (at 1o′clock; v3 in the Banff scoring system). There was no evidence of DSA, and a C4d stain was negative confirming the diagnosis of a TCMR process. The rejection episode responded to therapy. (PAS ×600 oil.) |
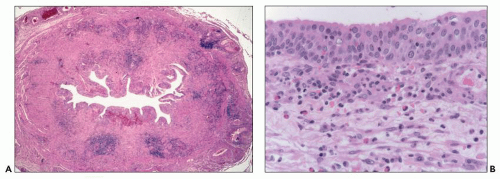 FIGURE 29.20 A: Ureter with acute cellular rejection. At low power, dense lymphocytic submucosal infiltrates are seen. (H&E ×25.) B: Urothelial invasion of mononuclear cells in acute rejection, involving the calyceal mucosa. Lymphocytes and eosinophils are in the submucosa. (H&E ×400.) |
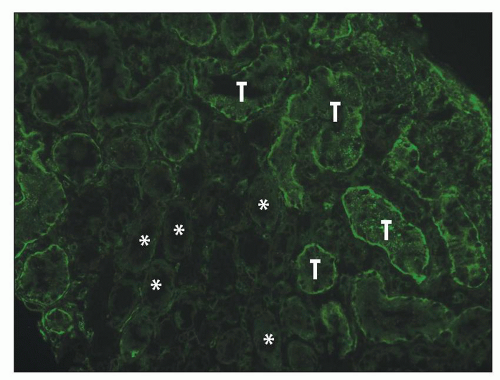 FIGURE 29.21 In cases of acute TCMR, the release of interferon-gamma stimulates the expression of MHC class II (HLA-DR) in the cytoplasm of tubular epithelial cells best detected by immunofluorescence staining (T, tubular cross sections with MHC class expression; asterisk, tubules without class II). The detection of tubular MHC class II can serve as an adjunct diagnostic parameter to establish a diagnosis of acute TCMR, in particular if Banff borderline changes (Banff category 3) are present or to confirm the concurrence of AMR and acute TCMR. (Direct immunofluorescence microscopy on frozen tissue with an antibody directed against MHC class II, ×200.) |
Polymorphonuclear leukocytes, platelets, and fibrin strands are occasionally seen. Mitotic figures can be detected but are usually rare. Glomerular endothelial cells are reactive, with a marked increase in cytoplasmic organelles (ribosomes, mitochondria, endoplasmic reticulum) and nuclei show open chromatin and prominent nucleoli (Fig. 29.22). The endothelial cells typically lose their fenestrations and are often separated from the GBM by a widened lamina rara interna. Rudimentary thin subendothelial new lamina densa formation can occasionally be seen as an early ultrastructural sign of evolving transplant glomerulopathy (220) scored in the Banff 2013 update as “cg1a” (see Fig. 29.22D). The mesangium has loose matrix and sometimes monocytes. Podocytes primarily those overlying segments with glomerulitis usually show foot process effacement.
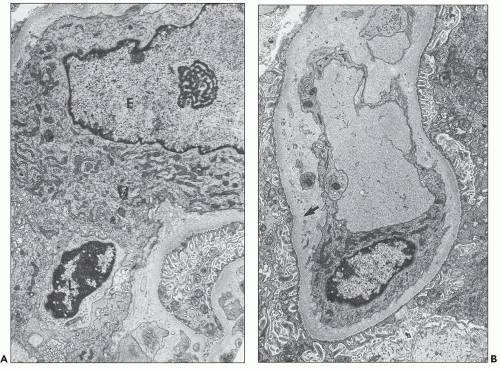 FIGURE 29.22 Transplant glomerulitis. A: A lymphocyte (L) is in contact with an activated glomerular endothelial cell (E). B: An activated endothelial cell has lost fenestrations and is separated from the original basement membrane (arrow) by an expanded subendothelial space/lamina rara interna that contains loose matrix, cell processes, and debris. |
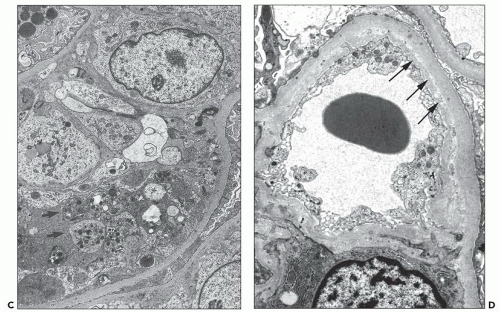 FIGURE 29.22 (Continued) C: Platelets are numerous in a capillary loop that has denuded endothelium. D: Activated endothelial cells are overlying a widened lamina rara interna containing rudimentary new densa as a sign of early, ultrastructural evidence of GBM remodeling and duplication (arrows). Light microscopy in this case showed focal minimal glomerulitis without transplant glomerulopathy. Early ultrastructural GBM remodeling/minimal duplication as illustrated in D is scored in the 2013 update of the Banff classification scheme as “cg1a”. (Electron micrographs. (A) ×6360, (B) ×5225, (C) ×6000, (D) ×5500.) |
features are specifically related to TCMR versus AMR has not been determined. Multilaminations of PTC basement membranes are typically minor but can become prominent with 5 to 7 basement membrane layers seen in 10% to 20% of late acute TCMR rejection episodes occurring post-year 1 (224). Such pronounced multilaminations in cases of acute TCMR (C4dnegative) represent protracted endothelial injury and evidence of early chronic rejection (224). The endarteritis lesions have been little studied ultrastructurally, because of the difficulty in sampling these very focal lesions.
Banff category 3 “borderline” changes, 33% of cases showed a molecular phenotype similar to TCMR, while 67% were nonrejection-like, underscoring on a molecular level the diagnostic heterogeneity of this group. The tightest association was between molecular phenotype and Banff total inflammatory (ti) score, indicating that the molecular phenotype is largely due to tubulointerstitial inflammation.
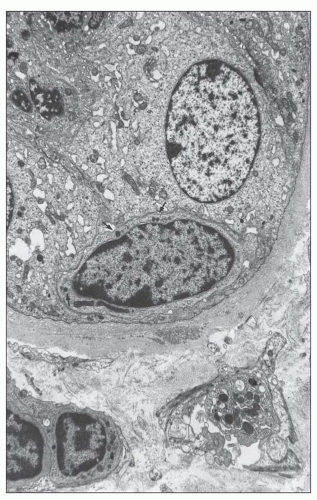 FIGURE 29.23 Acute cellular rejection. A tubule is invaded by a lymphocyte with a few dense cytotoxic granules (arrows) that are oriented toward the epithelial cells. The tubular basement membrane has fine laminations but does not appear disrupted. (Electron micrograph; ×5200.) |
IFN-γ, produced by antigen-activated T cells and NK cells, induces both class I and class II MHC antigens on epithelial and endothelial cells (263). Increased surface density of MHC molecules enhances the susceptibility to T-cell-mediated lysis and the ability to present antigen (264). However, induction of graft MHC molecules is not necessary to promote rejection. IFN-γ-deficient mice reject kidney allografts as quickly as normal mice without induction of MHC molecules (265). Paradoxically, class I- and class II-deficient grafts are also rejected efficiently, with MHC expression undetectable by immunoperoxidase techniques (266,267).
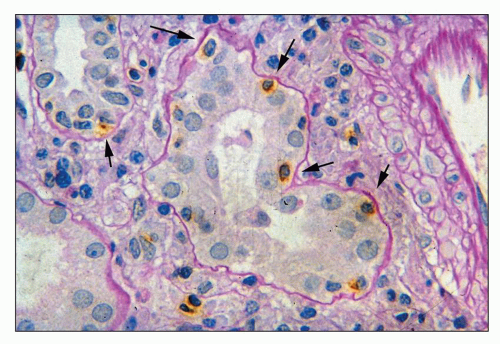 FIGURE 29.24 Acute tubulointerstitial cellular rejection (Banff category 4, type 1). Lymphocytes in tubules express the cytotoxic granule associated protein TIA-1 (GMP17). (PAS combined with anti-TIA-1 antibody/immunoperoxidase technique ×400.) |
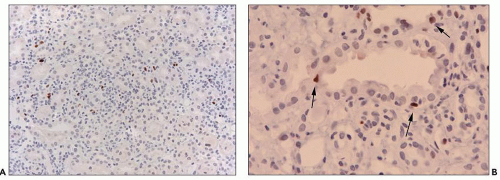 FIGURE 29.25 Acute tubulointerstitial cellular rejection (Banff category 4, type 1). A: Cells in the infiltrate stain for Foxp3, a transcription factor for T regulatory cells. B: Foxp3 cells are present in the tubules (arrows). (Anti-Foxp3 immunoperoxidase, (A): ×200, (B) ×600.) |
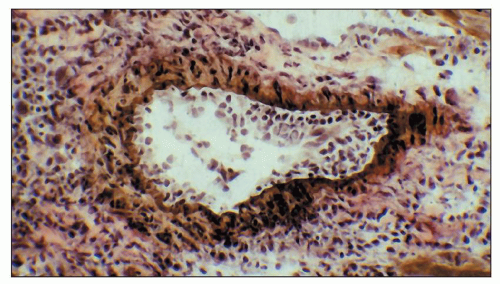 FIGURE 29.26 Endarteritis in a cardiac allograft in a mouse deficient in B cells. This shows that endarteritis lesions can occur without the participation of antibody (371). (Elastic tissue stain of a cryostat section, ×400.) |
antineutrophil cytoplasmic antibody (ANCA)-associated small-vessel vasculitis. In contrast to pyelonephritis, all these latter diagnoses typically lack dense intratubular polymorphonuclear cast material and polymorphonuclear leukocytes between injured tubular epithelial cells. Granulomas are extremely rare in allograft biopsies due to rejection alone, although they may occur in response to ruptured tubules. Granulomas have been associated with miliary tuberculosis, adenovirus, Escherichia coli urinary tract infection/pyelonephritis, and Candida albicans, generally with the organism demonstrable in the lesions (384). Drug allergy or sarcoidosis may also produce granulomatous interstitial nephritis.
appearance” of the intima. These changes are of no diagnostic and prognostic significance; they should not be misinterpreted as evidence of transplant endarteritis.
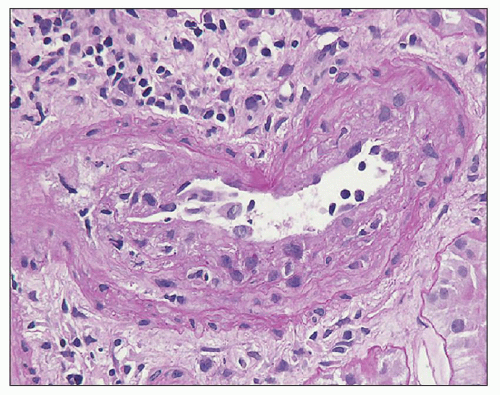 FIGURE 29.27 The differential diagnosis of endarteritis includes thrombotic microangiopathy (TMA) as illustrated in this case. The loose, thickened intima of TMA might contain rare mononuclear cells amidst abundant and dominating myofibroblasts that may resemble lymphocytes or macrophages at first glance. Clues that favor TMA over transplant endarteritis are insignificant inflammation, trapped and fragmented red blood cells, mucoid intimal changes, lack of C4d staining along PTC, and involvement of the glomerular vascular pole regions. (PAS ×400.) |
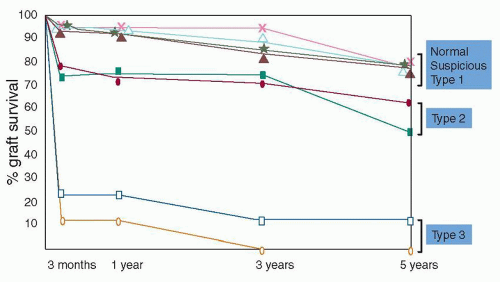 FIGURE 29.28 The Banff types of rejection correlate well with graft survival (206). Type I does not significantly affect prognosis, but both type II and especially type III do diminish graft survival. This study was done before C4d stains were widely used or routine posttransplantation DSA testing was performed. A recent single-center observation suggests that C4d/DSA negative type II rejection has a favorable prognosis (similar to type 1 rejection) compared to cases with a concurrent antibody-mediated rejection component (153). (Adapted from Bates WD, Davies DR, Welsh K, et al. An evaluation of the Banff classification of early renal allograft biopsies and correlation with outcome. Nephrol Dial Transplant 1999;14(10):2364-2369.) |
rejection episodes with arterial thrombus formation and the presence of DSA/C4d positivity. Occasionally, infarction can be associated with infections, especially CMV or productive adenovirus (see below) (401). Old infarcts are occasionally found in well-functioning grafts, dating from the time of transplantation; these are of no significance (402).
caliber vessels and is typically found as a sequela of transplant endarteritis (396). In sclerosing transplant arteriopathy, the intima usually contains varying numbers of myofibroblasts, occasional foam cells, and, in active disease stages, scattered, often clustered mononuclear inflammatory cells that can be most prominent along the inner elastic lamina. Potentially, even eosinophilic leukocytes play a role in the development of intimal sclerosis (414). Endothelial cells are often enlarged with reactive nuclei sometimes overlying an ill-defined ring of smooth muscle cells, that is, so-called neomedia formation (see Fig 29.29B and C). The inner elastic lamina usually remains intact without major breaks that are only prominent in cases with preceding transmural inflammation (Banff v3 lesions). The differential diagnosis of sclerosing arteriopathy includes TMAs (see Fig 29.27) and hypertension-induced arteriosclerosis characterized by marked arterial intimal fibroelastosis and a lack of intimal inflammation.
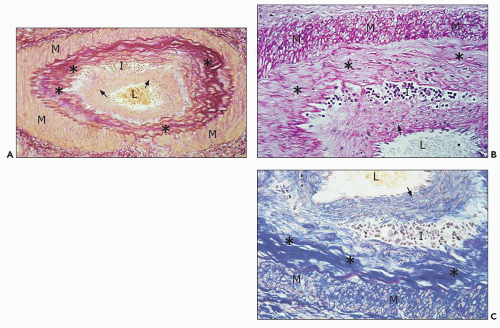 FIGURE 29.29 Chronic active TCMR with sclerosing transplant arteriopathy and lymphocytic infiltrates, C4d-negative, DSA-negative (Banff category 4 chronic TCMR). A: Five months after transplantation and 4.5 months after acute TCMR with transplant endarteritis (C4d-negative, DSA-negative; compare with Figure 29.15) that was superimposed on preexisting mild to moderate hypertension-induced donor-derived arteriosclerosis, a repeat biopsy shows an arcuate caliber artery with aggregates of inflammatory cells (I) in the widened intimal zone. Inflammation overlies donor-derived intimal elastosis (asterisk) and is located under a rim of chronic rejectioninduced de novo intimal sclerosis lacking elastosis (arrows). B, C: The subendothelial rim of chronic rejection-induced sclerosis shows on higher-power examination a vaguely organized pattern with a loose arrangement of myofibroblasts called “neomedia formation.” ((A) Elastic stain, ×200; (B) PAS, ×400; (C) trichrome, ×400; L, arterial lumen; M, medial smooth muscle layer.) |
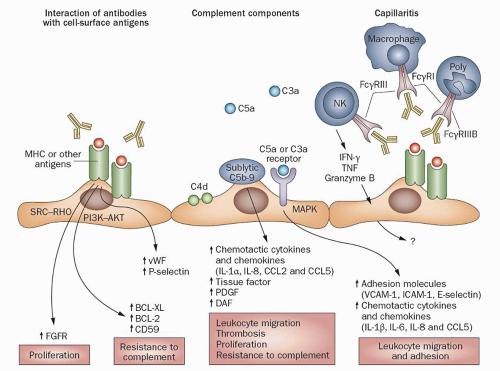 FIGURE 29.30 Three major pathways by which antibody can affect the endothelium. (1) Antibody to MHC acting alone in vitro on cultured endothelium promotes proliferation and activation of multiple signaling pathways. (2) Complement activation by DSA can attract inflammatory cells through C3a and C5a and cause lysis of endothelial cells (462) and expression of adhesion and procoagulant molecules. (3) DSA can potentially mediate endothelial injury and activation via Fc receptors on NK cells, monocytes, and granulocytes. (Reprinted from Farkash EA, Colvin RB. Pathology: Diagnostic challenges in chronic antibody-mediated rejection. Nat Rev Nephrol 2012;8(5):255-257.) |
In the older literature, about 85% to 90% of the patients with C4d+ had positive tests for circulating DSA, but these tests were less sensitive than current solid-phase assays. In recent protocol biopsies of presensitized patients, all biopsies with C4d had circulating DSA by solid-phase assay, arguing against the hypothesis that the kidney could absorb enough DSA to render this highly sensitive serologic test negative (480). The deposition of C4d in the absence of detectable DSA to MHC is strong presumptive evidence for non-MHC endothelial target antigens.
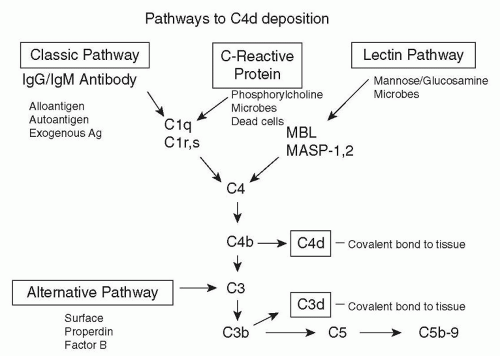 FIGURE 29.31 Pathways of C4 activation (389). Activation of C1 (composed of C1q, C1r, and C1s) is initiated by interaction of C1q with IgG or IgM bound to epitopes on the graft endothelium. C4 is cleaved by C1s into C4a and C4b, exposing a sulfhydryl group. The reactive sulfhydryl group of C4b rapidly forms an ester or amide bond with nearby molecules containing hydroxyl or amino groups. C4b combines with the enzymatically active fragment C2a to form C4bC2a, which is known as the classical pathway C3 convertase. C4bC2a cleaves C3 into C3a and C3b (which also has a reactive sulfhydryl group) and with the C3b molecule covalently deposited in the immediate vicinity and forms the C5 convertase C4bC2aC3b. Cleavage of C5 releases a bioactive peptide C5a and C5b. C5b initiates formation of the membrane attack complex (MAC; membrane-bound C5b-9), which causes cell lysis (435). The lectin pathway is stimulated when mannan-binding lectin (MBL), L-ficolin, or H-ficolin binds to the appropriate carbohydrate (typically on pathogens or apoptotic cells) (467). MBL binds to mannose (or glucosamine), and the ficolins bind to N-acetylglucosamine. L-ficolin also binds to elastin and lipoteichoic acid, and H-ficolin also binds to N-acetyl galactosamine. MBL, L-ficolin, and H-ficolin (all homologous to C1q and fibrinogen) activate C4 via their associated serine proteases, MASP-1 and MASP-2 (homologous to C1r and C1s). C4 is also activated via the binding of C-reactive protein (CRP) to the carbohydrate, phosphorylcholine with participation of C1q (1303). Little or no terminal components (C5b-9) are generated, because CRP simultaneously recruits Factor H, and may thus provide an anti-inflammatory effect. (From Rotman S, Collins AB, Colvin RB. C4d deposition in allografts: Current concepts and interpretation. Transplant Rev 2005;19:65.) |
of C4d deposition in PTC and had a lower reproducibility than IF (kappa 0.3 vs. 0.9, respectively) (487). Furthermore, there is considerable interinstitutional variability in IHC results, as shown by an international quality assurance project involving 73 centers (490). Some variation was attributed to techniques. Heat-induced epitope recovery (pH 6 to 7, 20 to 30 minutes, citrate buffer) with polyclonal antibody incubation (less than 1:80, greater than 40 minutes) appeared to be the best practice (490). Not uncommonly, the plasma in the capillaries is fixed by the formalin processing and also stains for C4d by IHC, which interferes with interpretation. Extravasation of C4d into the connective tissue is also common and should not be mistaken for capillary wall deposition. If extensive, these samples are not interpretable.
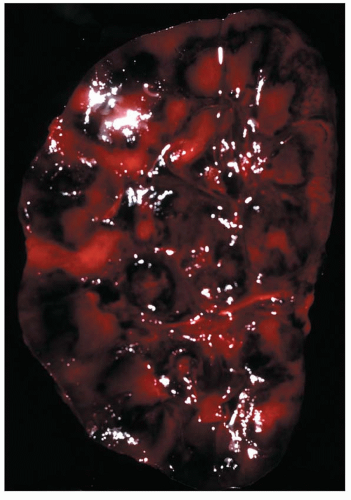 FIGURE 29.32 Hyperacute rejection, nephrectomy. The cut surface of the markedly swollen kidney is grossly hemorrhagic and glistening with edema fluid (hence the reflections). |
C4d positivity along PTC. Lack of C4d deposition in one case of hyperacute rejection was attributed to non-complement-fixing donor-specific antiendothelial antibodies (502).
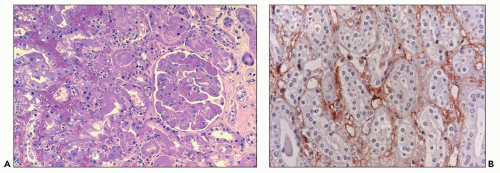 FIGURE 29.33 Hyperacute rejection due to preexisting antidonor class II HLA antibodies (509). A: Interstitial edema and hemorrhage are conspicuous as are neutrophils in PTC. Glomeruli are congested and have lost endothelial nuclei. (H&E, original magnification 200×.) B: C4d on paraffin section shows widespread, circumferential deposition along the PTC (stain done on an unstained paraffin section stored for 25 years). (C4d IHC, original magnification 400×.) |
the PTC in patients with antiendothelial antibodies (205,507). Preexisting donor kidney reactive antibodies were detected by immunoperoxidase techniques in 19% of 70 patients who had a negative T-cell cytotoxic crossmatch, and 50% reacted with endothelium (some also with epithelium) (509).
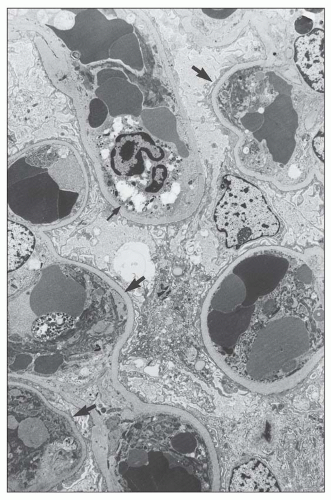 FIGURE 29.34 Hyperacute rejection due to anti-HLA-DR antibodies, biopsied at 24 hours after transplantation. Electron micrograph of glomerular capillaries shows fibrin platelet thrombi (thick arrows), a degranulated neutrophil (arrow), and compacted red cells; the endothelium is absent from most of the GBM. (×5000.) |
an episode of acute rejection or no immediate effect on graft function (accommodation). The long-term outcome of these recipients is unknown but of great interest.
TABLE 29.5 Conditions caused by antibody-induced graft injurya | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
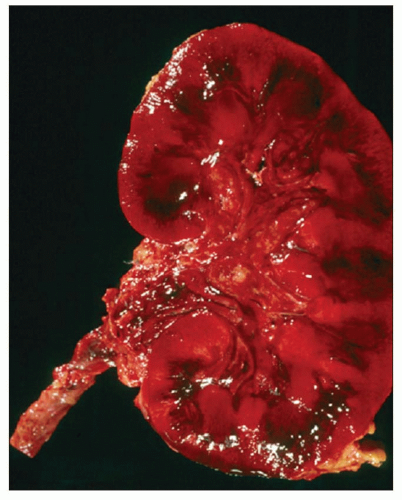 FIGURE 29.35 Gross appearance of an allograft with severe acute AMR 16 days after transplantation that failed after initially functioning for several days; ABOi graft (A2 into O). |
(387). Mesangial IgM and IgG may be more prominent than in non-AMR, but the difference (43% vs. 17%) is not diagnostically useful (169).
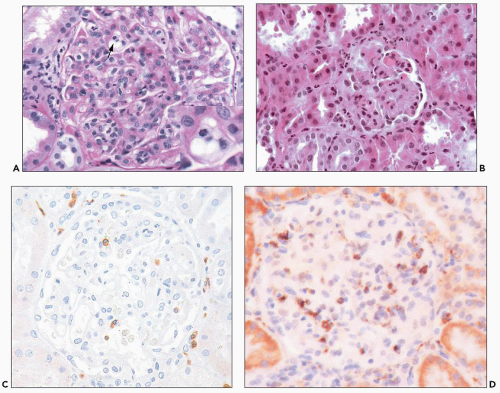 FIGURE 29.36 Acute AMR with glomerular inflammation (compare to Figure 29.7). A: Transplant glomerulitis with mononuclear cells, neutrophils, and reactive endothelial cells. An endothelial mitosis is shown (arrow, insert) C4d stain along PTC was positive. (PAS stain, original magnification 400×.) B: Transplant glomerulitis with neutrophils and thrombi. (H&E original magnification 20×.) C: Transplant glomerulitis in acute AMR with a few CD3+ cells (T cells). (Immunoperoxidase stain.) D: Same graft showing many CD68 cells (macrophages). The graft had prominent C4d deposition along PTC. (Immunoperoxidase stain.) (C, D courtesy of Alex Magil, Vancouver.) |
the frequency is uncertain. In a series with serial biopsies of presensitized patients, C4d became positive later after the capillaritis had been present for several days (549).
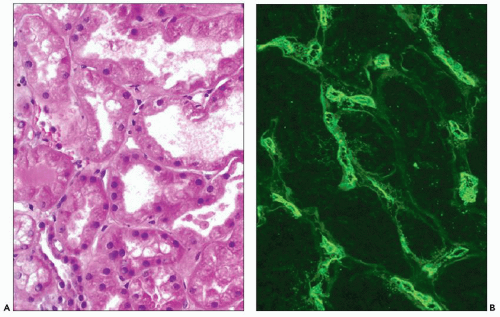 FIGURE 29.37 Acute AMR present on day 9 of DGF. A: Pattern of acute tubular injury, without evidence of inflammation. (H&E stain, original magnification 400×.) B: Immunofluorescence C4d stain on cryostat section shows widespread staining of PTC. (Original magnification, 400×.) |
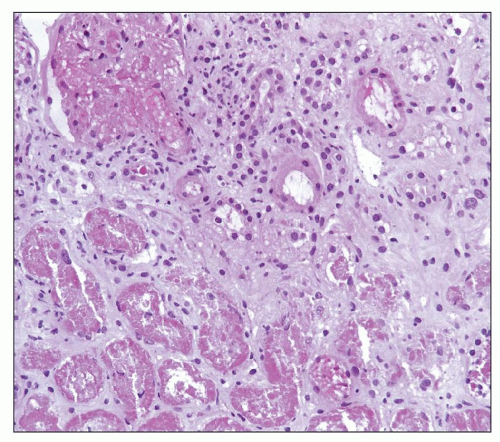 FIGURE 29.38 Acute AMR with glomerular and tubular necrosis. (H&E original magnification 200×.) |
flow crossmatch at the time of transplantation (118). In 1-week protocol kidney biopsies, Sund showed PTC endothelial C4d deposition in 30% of cases; 33% of these did not meet histologic criteria of acute rejection, but 82% developed rejection during further follow-up. Koo reported C4d in 13% of 48 one-week protocol biopsies (556). C4d was present in 33% of samples with rejection and 3% of samples without rejection; all 5 with C4d and rejection had DSA (556). Outcome at 1 year was not affected by C4d status at 7 days, despite the lack of specific treatment.
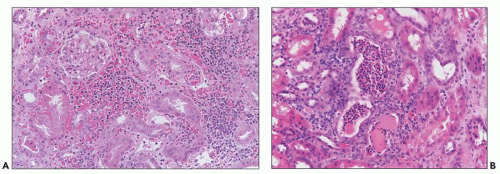 FIGURE 29.39 Acute AMR (A) Widespread interstitial hemorrhage is present. B: Neutrophils are in tubules, resembling acute pyelonephritis. (H&E, original magnification 400× (A) and 200× (B).) |
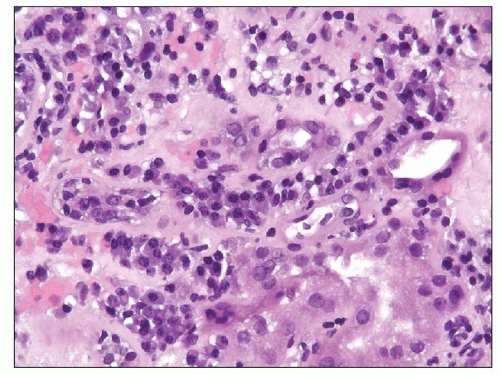 FIGURE 29.40 Plasma cell-rich late acute TCMR, C4d-positive (compare to Figure 29.13). The combination of plasma cell infiltrates, that is, acute plasma cell-rich TCMR and acute AMR, is a very rare occurrence that seems to carry a poor prognosis. (H&E original magnification 400×.) |
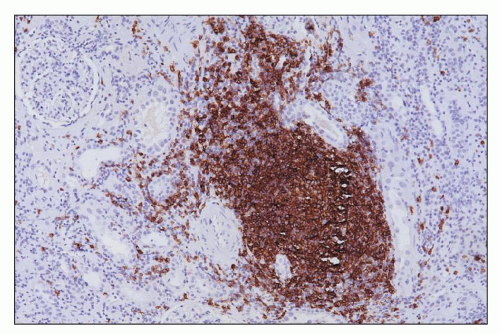 FIGURE 29.41 Acute AMR with a nodule of B cells, a finding that has been reported by some to carry an adverse prognostic significance. At present, there is no linkage between B cells in the interstitial infiltrate and the presence of antidonor antibodies. (Immunohistochemical stain for CD20, original magnification 100×.) |
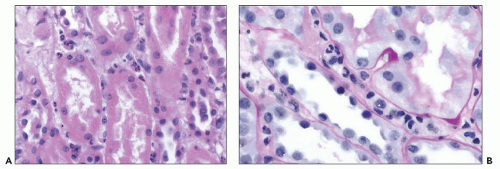 FIGURE 29.42 Acute AMR with characteristic intraluminal PTC leukocytes. Primarily neutrophils (A) and primarily mononuclear (B) cells. (H&E (A) and PAS (B). Original magnification 400× (A) and 600× (B).) |
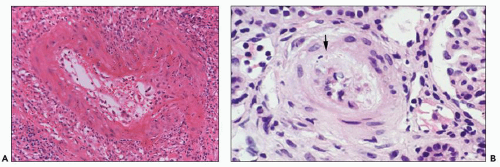 FIGURE 29.43 Acute AMR (A) Fibrinoid arterial necrosis (Banff type III). Neutrophils and fibrin are seen in the wall of the arcuate sized artery. Nephrectomy specimen. (C4d stain-positive. H&E original magnification 200×.) B: Mucoid intimal thickening resembling TMA. Patient had anti-class I antibodies (134). Red cell fragment in intima (arrow). (H&E, original magnification unknown.) (Courtesy of Kim Solez.) |
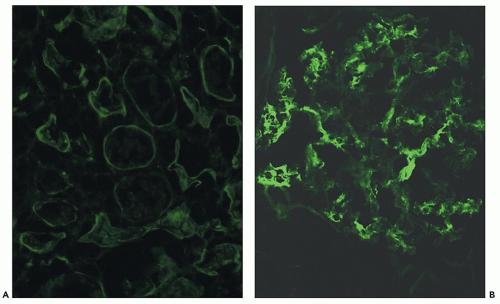 FIGURE 29.44 Normal kidneys stained for C4d using a monoclonal antibody cryostatin cryostat sections. A: Negative PTC and faint staining along the TBM. B: Prominent mesangial deposits are present in normal glomeruli. (Original magnifications 400×.) |
as 7 to 8 days after a positive biopsy (150) and in rat heart transplants in 5 days (557).
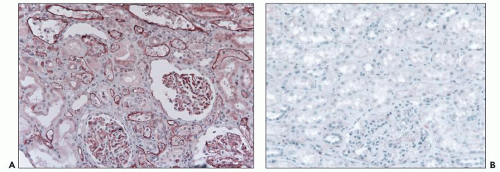 FIGURE 29.45 Paraffin sections stained using a polyclonal anti-C4d (347). A: Acute AMR showing widespread, circumferential deposits in characteristically dilated PTC containing leukocytes. The glomerular capillary walls also stain prominently. B: Normal kidney stained with the same technique demonstrates the absence of C4d in the glomeruli in paraffin sections (compare with Fig. 28-45B). (Original magnification 200×.) |
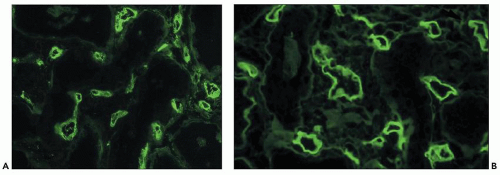 FIGURE 29.46 Acute AMR, C4d stain using monoclonal antibody to C4d in cryostat sections. Bright widespread, circumferential staining is present in (A) and (B). A: Has a slightly granular appearance, and (B) is purely linear. Both patients had antibodies to donor HLA antigens. (Original magnification 400×.) |
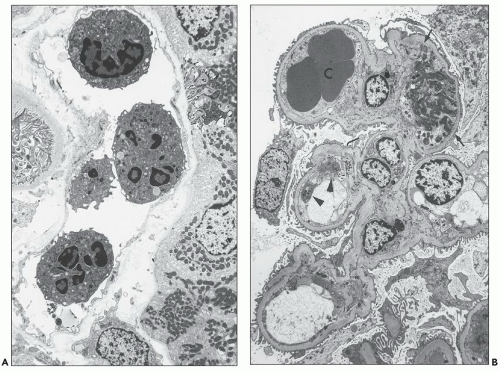 FIGURE 29.47 Acute AMR. A: Neutrophils are in PTC whose endothelium shows the subtle changes of injury (loss of fenestrations). B: A glomerulus has one capillary plugged with fibrin (arrows) and another filled with compacted red cells surrounded by a reactive endothelial cell (C ); a third loop has a few platelets (arrowheads). (Electron micrographs. (A) ×3,700; (B) ×3,300.) |
AMR. A negative C4d stain or a negative serology does not rule out acute AMR. Histology by itself is insufficient to rule acute AMR in or out, and the diagnosis can be complicated by other pathology, such as concurrent acute TCMR (82,142,150,562).
| ||||||||||||
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Glomerular Diseases With Organized Deposits
Glomerular Diseases With Organized Deposits
 Renal Changes With Aging and End-Stage Renal Disease
Renal Changes With Aging and End-Stage Renal Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


