glomerulopathy and also discusses other glomerular diseases with organized deposits that are covered in more detail in other chapters (e.g., cryoglobulinemic glomerulonephritis and collagenofibrotic glomerulopathy). In addition, normal extracellular materials that can be confused with organized deposits (e.g., fibrillary collagen and fibrin tactoids) are described. The organized deposits discussed in this chapter are the ones that are seen most often in renal biopsy specimens (8,9,10); however, there are anecdotal reports of organized deposits that do not fit well into these recognized categories (11).
TABLE 23.1 Congo red reactivity of glomerular deposits with ultrastructural organized deposits | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
present in 50% of patients at the time of biopsy (16), and 65% are hypertensive (16). In the two most recent and largest series (18,19), associated medical conditions included, respectively, diabetes mellitus (20%, 20%), lymphoproliferative disease (2%, 9%), carcinomas (5%, 14%), and systemic lupus erythematosus (1.6%, 3%). In the series from the Mayo Clinic (19), an additional 15% had miscellaneous autoimmune diseases. Positive serologic tests in the same two series included hepatitis C (17%, 3%), monoclonal immunoglobulin by serum or urine protein electrophoresis (15%, 17%), antinuclear antibodies (16%, 14%), low complement level (2%, 2%), and cryoglobulins (4%, 3%). FGN is a disease confined to the kidney. There are individual case reports indicating involvement of the liver, lungs, and heart (22,23,24); however, careful examination of the published illustrations suggests to us as well as to others (25) that interpretation of these reports warrants caution. An autopsy study appears to have demonstrated the presence of the characteristic fibrils of FGN only in the spleen in addition to the kidney and confirmed that localization by the immunogold labeling method (26).
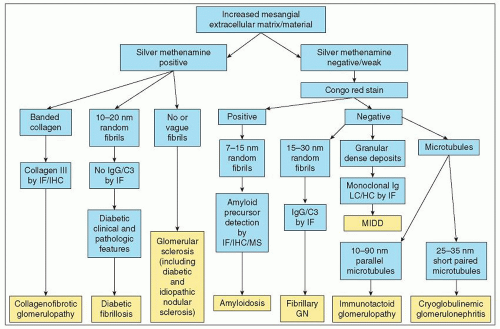 FIGURE 23.1 Diagnostic algorithm for glomerular mesangial extracellular matrix expansion based on silver methenamine staining. Many but not all processes with organized deposits are included. Rare entities such as fibronectin glomerulopathy and metabolic storage diseases are not included. MIDD, monoclonal immunoglobulin (Ig) deposition disease, including light chain (LC) and heavy chain (HC) deposition disease; GN,glomerulonephritis; IF, immunofluorescence microscopy; IHC, immunohistochemistry; MS, mass spectroscopy. |
The above-described mesangial pattern comprised 71% of cases in a large series reported from the Mayo Clinic (19); however, other patterns described (18) include membranoproliferative-like, diffuse proliferative and exudative, segmental necrotizing and crescentic, membranous and diffuse sclerosing patterns. Crescents are reported to be present in between 17% and 31% of cases (12,18,19,20) excluding the category of segmental necrotizing and crescentic glomerulonephritis, which was defined by Nasr et al. (19) as presence of ≥50% glomeruli with segmental tuft necrosis or crescents.
TABLE 23.2 Distinguishing pathologic characteristics of different types of organized glomerular deposits | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
and may simulate the appearance of anti-glomerular basement membrane autoantibody disease (27). The appearance of immunofluorescence in FGN is particular to the entity and allows one to anticipate the diagnosis in the great majority of cases even before electron microscopic examination, although of course by definition ultrastructural examination is required to render a confident diagnosis of FGN. Although the signal of staining for IgG is usually strong, it is neither granular nor linear and has been described as smudged (28) (Fig. 23.5). There is a clear predilection for staining with subclass IgG4 first demonstrated in 13 cases studied by Iskandar et al. (12) with only weaker staining in 15% of cases for subclass IgG1. This has generally been confirmed in a subsequent series (18). A single case of IgA-λ restriction has been reported (29). In two cases that appear to be bona fide FGN based on the published electron micrographs, attempts at immunofluorescence staining with antihuman immunoglobulin (Ig) antibodies from multiple commercial suppliers failed to demonstrate the presence of Ig deposits (30).
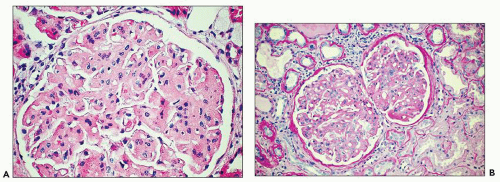 FIGURE 23.2 Fibrillary glomerulonephritis. A: Representative glomerulus from a case of FGN with extensive mesangial expansion. Note the infiltration of the mesangium by a homogeneous though slightly “moth-eaten” eosinophilic material. (H&E, 400×.) B: Case of FGN displaying a diffuse proliferative pattern and thickening of capillary walls. Note that the infiltrating material is PAS reactive. (PAS stain, 200×.) |
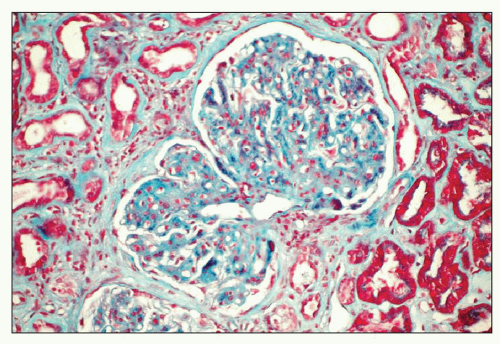 FIGURE 23.3 Fibrillary glomerulonephritis. Same case as in Figure 23.2 using Gomori trichrome green stain for collagen, showing the increased extracellular matrices staining green with various degrees of segmental fuchsinophilic tinge. (Gomori trichrome, 200×.) |
deviation of 7.4 nm (12) (Fig. 23.6). The fibrils are often set in a background of amorphous immune complex-type electron-dense deposits. Occasionally, the fibrillary deposits infiltrate the glomerular basement membrane and produce perpendicular spicular projections similar to those seen in amyloidosis. In rare cases corresponding to a membranous pattern by light microscopy, subepithelial deposits are present with little or no infiltration of the glomerular basement membrane seen in most cases (18,19). Tubular basement membrane deposits are very rarely present (18,19,20,31).
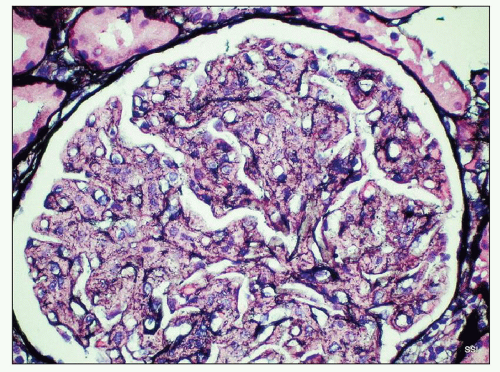 FIGURE 23.4 Fibrillary glomerulonephritis. Periodic acid-Schiff methenamine silver (PAMS/Jones) stain showing markedly reduced staining in the expanded mesangium and the thickened capillary walls. Staining is seen only in the residual GBM and remnants of fibrils of the mesangial matrix. (PAMS stain, 400×.) |
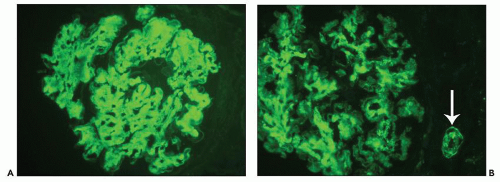 FIGURE 23.5 Fibrillary glomerulonephritis. A, B: Glomeruli illustrate the characteristic strong smudgy mesangial and capillary wall staining characteristic of FGN. B: Rare smudgy staining of the hilar arteriole (arrow). (Direct immunofluorescence, fluorescein isothiocyanate-labeled rabbit anti-human IgG, 400×.) |
however, this is an appropriate place to mention that of the long list of amyloidogenic proteins provided in Table 23.1, the ones likely by far to involve the kidney are AL amyloidosis and AA amyloidosis although recently cases of leukocyte chemotactic factor 2 amyloid have been reported with increasing frequency (32) in particular in patients of Latino heritage. Leukocyte chemotactic factor 2 amyloid has a striking tendency to involve the interstitial compartment although the glomeruli and extraglomerular blood vessels are not spared. Most other forms of amyloidosis rarely involve the kidneys.
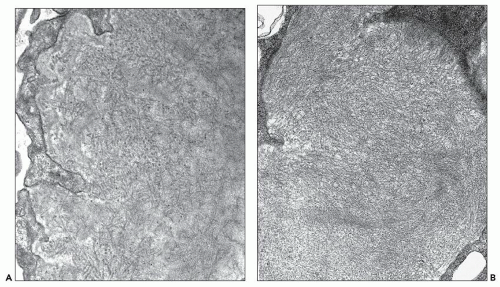 FIGURE 23.6 Fibrillary glomerulonephritis and amyloidosis. A: Electron micrograph of FGN deposit illustrating the randomly disposed nonbranching fibrils resembling amyloid fibrils but noticeably thicker. B: Electron micrograph of amyloidosis to contrast the thickness of the fibrils with those of fibrillary glomerulonephritis. (Uranyl acetate and lead citrate, 25,000×.) |
the immunoglobulins in a manner analogous to tactoid formation in the case of sickle cell disease.
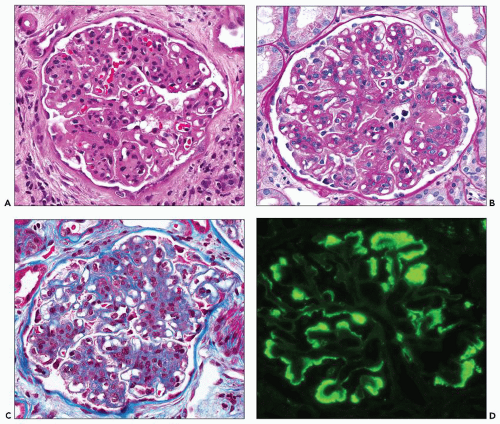 FIGURE 23.7 Monoclonal IgG lambda immunotactoid glomerulopathy with a membranoproliferative pattern with mesangial matrix expansion and hypercellularity, thick capillary walls, and slight lobulation A: H&E stain; B: PAS stain; C: Masson trichrome stain; D: Immunofluorescence microscopy showing capillary wall and mesangial staining for lambda light chains. Staining for IgG was similar, and staining for kappa light chains was negative. (400×.) (Courtesy of Carlos Andres Jimenez Guerrero and Adil Mohamed Hussein Gasim, Chapel Hill, NC.) |
infiltrating microtubular material, the glomeruli may show focal segmental hyalinosis, and sometimes, intraluminal hyaline thrombi are present, representing aggregates of microtubules. None of the 6 cases reported by Fogo et al. (20) and Rosenstock et al. (18) demonstrated crescents, 2 of the 14 cases reported by Bridoux et al. (17) had crescents, and none of the 16 cases reported by the Mayo Clinic group had crescents (41). The trichrome-stained sections present an appearance similar to that described in connection with FGN. The sections stained with Jones stain are described as showing a moth-eaten appearance because of failure of the mesangium to pick up the silver stain. Of course, the stains for amyloid are negative.
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


