specific term primary PLA2R-associated MGN (5). While the majority of cases represent the primary disease, MGN may occur secondary to many other conditions. With some exceptions, the pathology of primary and secondary MGN is often similar or even identical; thus, the diagnosis of primary MGN requires careful exclusion of the known secondary etiologies. In a review of nine series published between 1975 and 1989, Glassock (6) found that 23% of cases of MGN were secondary with the prevalence higher (35%) in children younger than 16 years and adults older than 60 years as compared to adults from 16 to 60 years of age (20%). The most common secondary etiology of MGN is systemic lupus erythematosus (SLE; i.e., membranous lupus nephritis [LN]). Most cases of secondary MGN relate to autoimmune/collagen vascular disease, infection, neoplasia, or therapeutic agents (i.e., drug-induced MGN) (6). New associations continue to appear, and critical evaluation is necessary before accepting an etiologic relationship. The well-established secondary etiologies of MGN covered in this chapter include therapeutic agents (gold, penicillamine, mercury, captopril, and nonsteroidal anti-inflammatory drugs [NSAIDs]), malignancy, infections (hepatitis B virus [HBV], hepatitis C virus [HCV], and syphilis), and miscellaneous conditions (sarcoidosis, IgG4-related disease [IgG4RD], allogeneic transplantation, and autoimmune thyroiditis). Chapter 14 covers MGN associated with SLE and other autoimmune/collagen vascular disease. Primary MGN is the focus of this chapter, with an understanding that it is critical to exclude secondary etiologies due to their different prognostic and therapeutic implications.
TABLE 7.1 Best established secondary etiologies of MGN | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
much less common etiology of nephrotic syndrome in children, accounting for only 3% of renal biopsies in a recent large pediatric biopsy series (23). Secondary forms of MGN are more common in the pediatric population (6) and most often relate to SLE (14) or infection, in particular HBV (13). At the opposite extreme, MGN is the most common etiology of nephrotic syndrome among patients above the age of 60, accounting for 32% of cases (24). This also holds true when only considering patients above the age of 80 (25). There is also significant experience with MGN in pregnant women who often experience a doubling of 24-hour urine protein, new onset of hypertension and, in some cases, renal insufficiency (26). The hypertension and worsening proteinuria often but not always reverse following parturition (26).
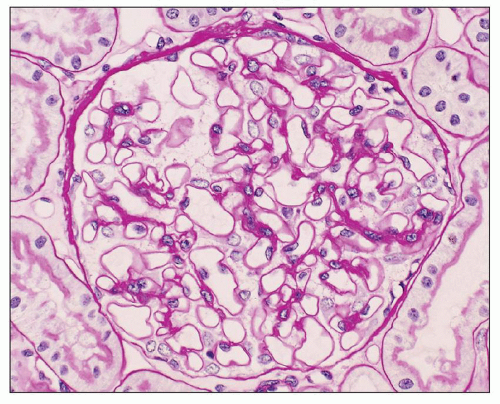 FIGURE 7.1 Histopathology of early MGN (stage I). The glomerulus exhibits no apparent abnormalities by light microscopy. The glomerular basement membrane appears thin and delicate, without evidence of thickening or spike formation. (PAS, ×200.) |
not an optimal stain for demonstration of GBM pathology because it does not distinguish well between the cell cytoplasm, immune deposits, and extracellular matrix. The GBM changes in MGN are best appreciated using the JMS stain (27) but are also well visualized with the PAS stain. By light microscopy, the earliest change in MGN is mottling of and small depressions in the GBM seen in en face sections stained with JMS (Fig. 7.5). This appearance is subtle and results from alterations in the GBM caused by the subepithelial deposits. More commonly, the JMS stain will reveal further developed changes of MGN represented by small projections (spikes) of silver-positive material (Fig. 7.6) composed of type IV collagen and noncollagenous extracellular matrix proteins, including laminin, heparan sulfate proteoglycan, and vitronectin (28,29,30). Initially, the spikes may be small and segmental, and a careful search under oil immersion is necessary to demonstrate them. Cases such as this, with only focal spikes, may represent either an earlier or a milder form of the disease (15), but immune deposits in cases with segmental spikes usually are more diffusely distributed than indicated by the light microscopic changes. As the lesion progresses, the spikes become larger, thicker, and diffuse (Fig. 7.7). Eventually, the GBM becomes more prominently expanded by a thick band of argyrophilic material containing abundant nonargyrophilic “holes” and imparting a vacuolated appearance (Fig. 7.8). The “holes” represent a normal-thickness GBM beneath the deposits surrounded by a heaped-up matrix around the deposits. In many cases, combinations of these various abnormalities including spikes, holes, and complex thickening of the GBM are present simultaneously in the same biopsy. The nonargyrophilic material deposited within the GBM and on its epithelial side stains red with the trichrome stain, while the surrounding GBM stains blue or green depending on the method used (Fig 7.9). The deposits
also stain blue in toluidine blue-stained 1-µm sections of plastic-embedded tissue. However, both the trichrome and toluidine blue demonstration of deposits are inconsistent, and these stains may be negative even in obvious cases of MGN. Thus, the more consistent histologic changes in MGN are thickening and remodeling of the GBM itself, seen best with the JMS and, to a lesser extent, PAS stains.
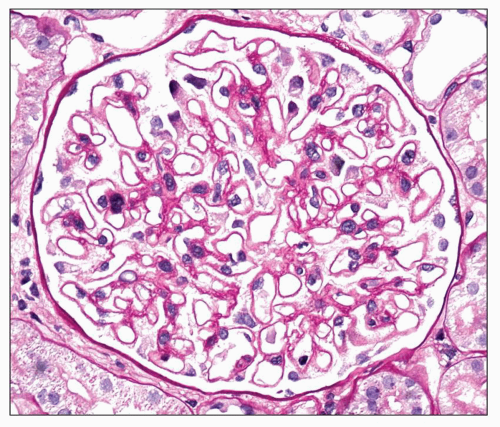 FIGURE 7.2 Histopathology of MGN with mild glomerular basement membrane (GBM) thickening (stage II). The GBM exhibits mild diffuse thickening with spike formation. Diffuse swelling of visceral epithelial cells is noted. (PAS, ×400.) |
 FIGURE 7.3 Histopathology of GBM thickening in MGN. There is marked global thickening of the glomerular basement membrane, which has a vacuolated appearance. (PAS, ×400.) |
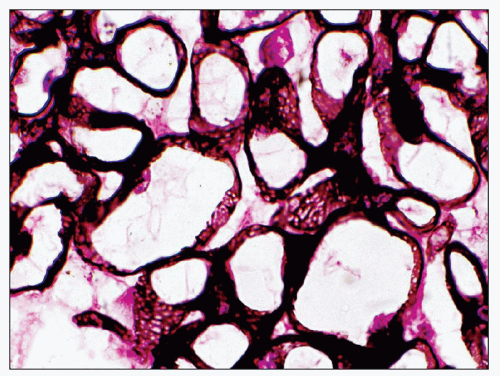 FIGURE 7.4 Histopathology of MGN. With the hematoxylin and eosin stain, the GBM has a thick and rigid appearance in this example of stage III MGN. Mild diffuse mesangial sclerosis is present. (H&E, ×400.) |
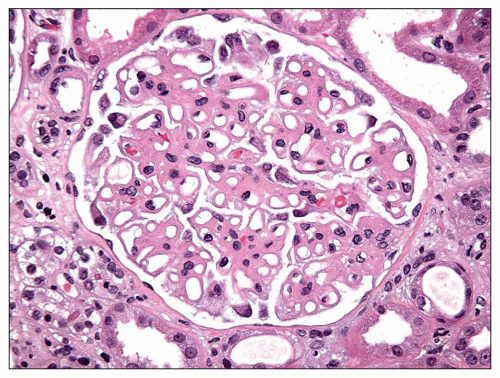 FIGURE 7.5 Histopathology of stage I MGN. In stage I MGN, the JMS stain may demonstrate small holes or depressions in the GBM. (JMS, ×600.) |
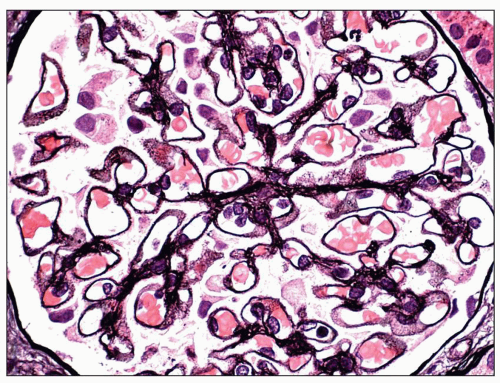 FIGURE 7.6 Histopathology of early stage II MGN. In early stage II MGN, the JMS stain demonstrates short GBM spikes. (JMS, ×400.) |
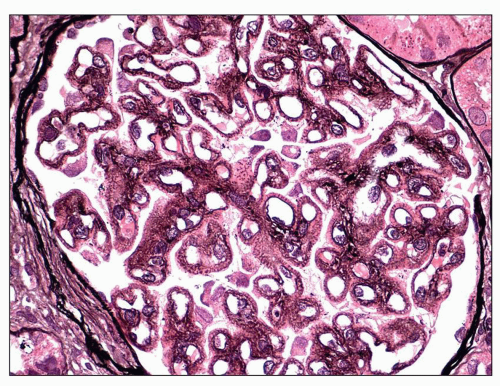 FIGURE 7.7 Histopathology of stage II MGN. In this better developed example of stage II MGN, the JMS stain exhibits more prominent GBM spike formation with intervening silver-negative subepithelial deposits (JMS, ×400.) |
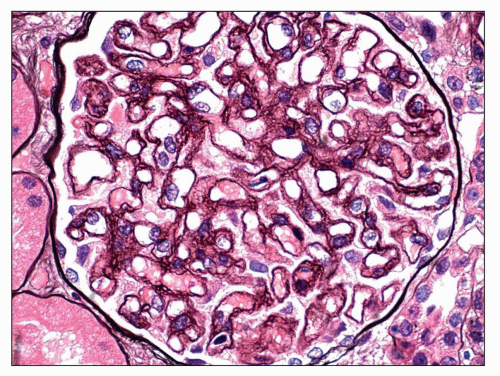 FIGURE 7.8 Histopathology of stage III MGN. In stage III MGN, the JMS stain reveals a globally vacuolated glomerular basement membrane (GBM) due to the presence of GBM spikes and overlying neomembrane formation. (JMS, ×400.) |
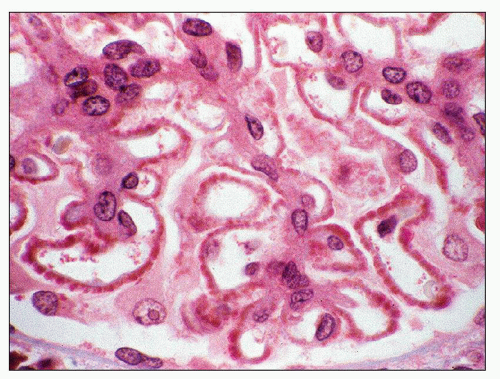 FIGURE 7.9 Light microscopy of subepithelial deposits in MGN. The Masson trichrome stain demonstrates fuchsinophilic deposits along the subepithelial aspect of the glomerular basement membrane. (Masson trichrome stain, ×600.) |
of coexistent IgAN (34,35). A secondary form of MGN (or coexistent IgAN) is particularly likely when the mesangial hypercellularity is accompanied by mesangial immune deposits (32,33,34,35,36).
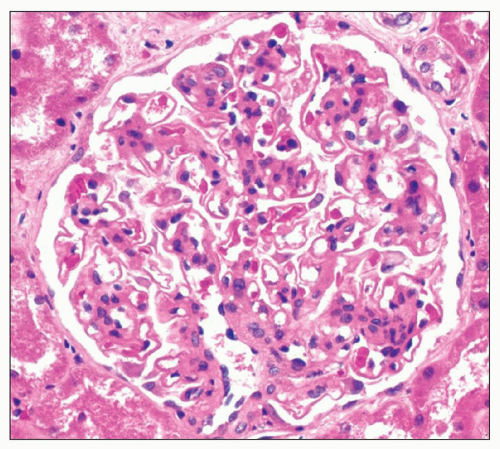 FIGURE 7.10 Mesangial hypercellularity in MGN. In this example of MGN, glomerular basement membrane thickening is accompanied by diffuse mesangial hypercellularity with up to 12 cells per mesangial area. (H&E, ×400.) |
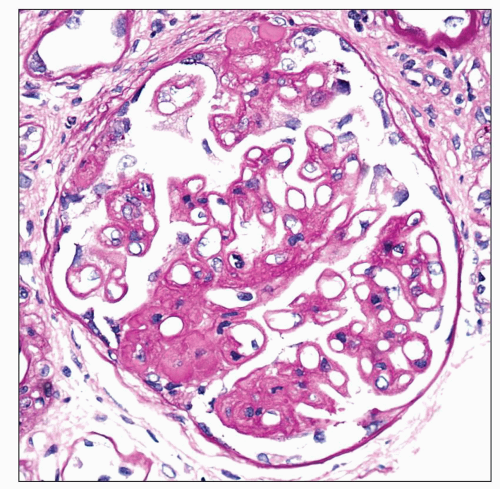 FIGURE 7.11 Focal segmental glomerulosclerosis in MGN. Findings of MGN are accompanied by two discrete lesions of segmental sclerosis with capsular adhesion. (PAS, ×400.) |
with the 24-hour urine protein excretion, serum albumin, and the percent of glomeruli with visceral epithelial cell protein resorption droplets, leading him to conclude that proteinuria per se played a role in the development of tubulointerstitial injury. A discussion of the role of proteinuria in the pathogenesis of tubular atrophy and interstitial fibrosis is included in the section on Etiology and Pathogenesis.
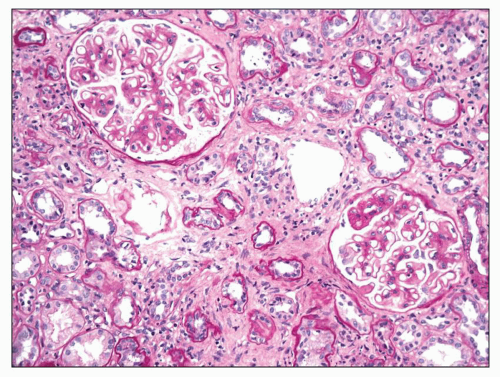 FIGURE 7.12 Interstitial fibrosis and tubular atrophy in MGN. In this example of MGN, the majority of tubules appear atrophic and are accompanied by diffuse interstitial fibrosis. (PAS, ×200.) |
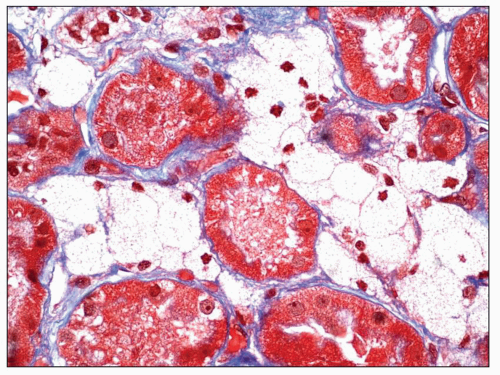 FIGURE 7.13 Interstitial foam cells in MGN. Interstitial foam cells expand the interstitium between tubules. (Trichrome stain, ×600.) |
Not surprisingly, these cases can be associated with hematologic malignancy (54,55).
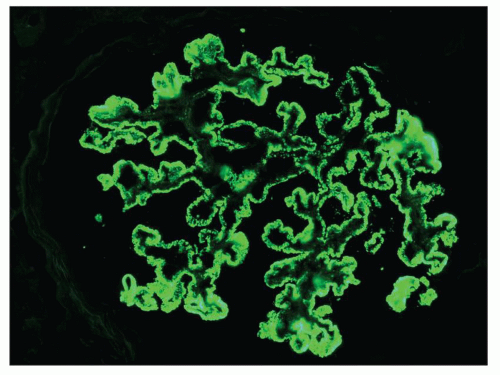 FIGURE 7.14 Immunofluorescence staining in MGN. Staining for IgG reveals intense, granular global subepithelial positivity involving the glomerular capillary walls. (×400.) |
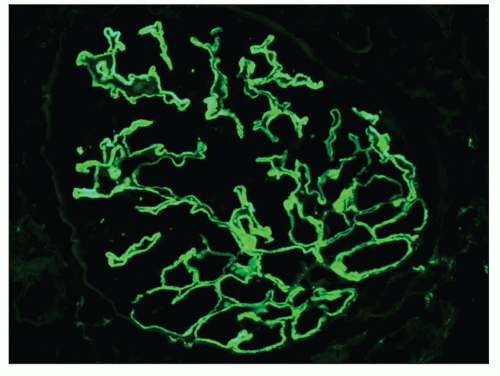 FIGURE 7.15 Immunofluorescence staining for IgG in early MGN. In early MGN, the subepithelial deposits may appear small and confluent, imparting a pseudolinear appearance. (IgG, ×400.) |
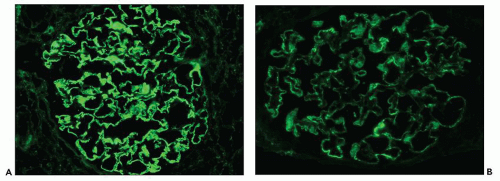 FIGURE 7.16 Comparative immunofluorescence findings in MGN. A: Staining for IgG reveals intense granular global positivity involving the glomerular basement membranes. B: In the same biopsy, a similar distribution but less intense staining for C3 is present along the GBM. (A and B: ×400.) |
IgG, for instance IgG1 (64). This possibility is supported by the observation that C3 is present in the subepithelial deposits in 85% of cases of MGN (33), a finding that cannot be explained by the presence of IgG4 alone, unless it is capable of activating complement through the lectin pathway (64,65). A recent study reconfirmed the dominance of IgG4 in primary MGN but noted that IgG1 predominated in early, stage I disease, suggesting that an IgG subclass switch occurs early in the course of primary MGN (66).
TABLE 7.2 Staining for IgG subtypes in primary and secondary forms of MGN | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
anti-TIN antigen has not been shown to be expressed in podocytes, although possible cross-reactivity of anti-TBM antibody with a podocyte antigen has been proposed (77). Prognosis for this condition is poor, with frequent progression to end-stage renal disease (ESRD).
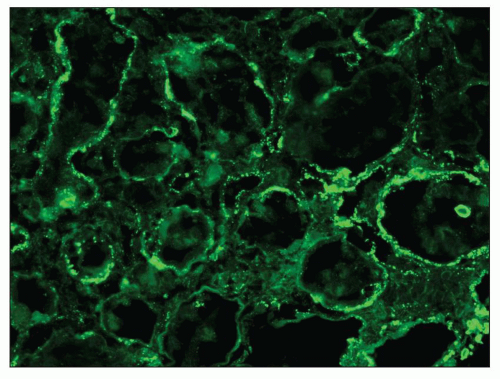 FIGURE 7.17 Tubular basement membrane deposits in MGN. In this patient with MGN, immunofluorescence reveals granular immune deposits within tubular basement membranes. The patient was subsequently found to have systemic lupus erythematosus. (IgG, ×400.) |
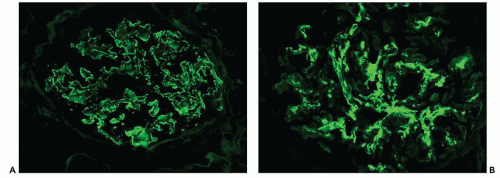 FIGURE 7.18 MGN with IgA nephropathy. A: Staining for IgG reveals granular global glomerular capillary wall positivity in a subepithelial distribution. B: Staining for IgA in the same biopsy reveals abundant mesangial deposits that are confined to the mesangium and do not appear to involve the peripheral capillary walls. (A and B: ×400.) |
deposits typically stain intensely by immunofluorescence. The three-dimensional appearance of the deposits can be partially appreciated by scanning EM of acellular glomeruli, where the elongated deposits are surrounded by a complex anastomosing network of basement membrane projections (84) (Fig. 7.23).
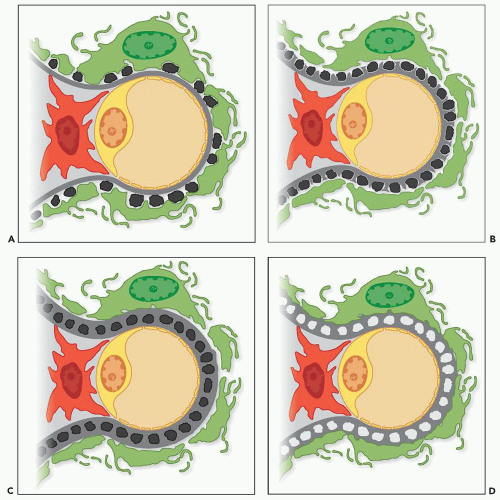 FIGURE 7.19 Depiction of the four stages of MGN as described by Ehrenreich and Churg. A: Stage I MGN is characterized by interspersed subepithelial deposits without intervening GBM spike formation. B: In stage II MGN, the subepithelial deposits are more numerous and more closely approximated with intervening GBM spikes. C: In stage III MGN, the GBM spikes extend over and encircle the deposits, incorporating them into the glomerular capillary walls. D: In stage IV MGN, the deposits lie within a mottled, irregularly thickened GBM and appear electron lucent, consistent with resorption. The irregular thickness of the GBM is consistent with extracellular matrix remodeling. |
deposits have a more electron-lucent appearance. By light microscopy, GBM thickening is most pronounced in stage III, and the JMS stain typically shows a complex, vacuolated GBM, while the deposits usually are not visible with the trichrome stain. Although the staining intensity may be reduced, the deposits are usually demonstrable by immunofluorescence. The findings of pure stage III MGN suggest a single generation of deposits, presumably formed over a similar time period. In contrast, most cases of stage III MGN have a mixed appearance with fresh deposits layered over older intramembranous deposits, suggesting a dynamic process of ongoing deposit formation, incorporation, and resorption. In such cases, it may be possible to assign a dominant stage based on the major pattern present. In many cases, two stages coexist, making it most appropriate to designate both stages (such as mixed stage II to III) (Fig. 7.25). By scanning EM, the complex network of GBM projections seen in stage II MGN is replaced by bridged segments of the neomembrane in stage III that have a relatively smooth appearance (84) (Fig. 7.26).
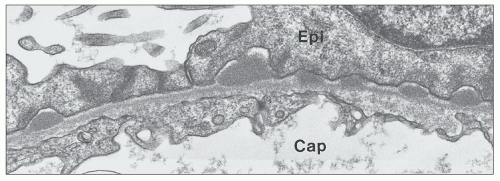 FIGURE 7.20 Electron micrograph of stage I MGN. In stage I MGN, there are small, interspersed subepithelial deposits without significant GBM spike formation. (×20,000.) |
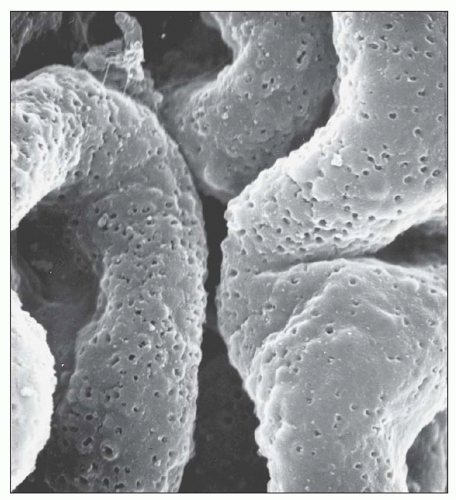 FIGURE 7.21 Scanning electron micrograph of an acellular glomerulus from a patient with stage I MGN. The cells were removed from the tissue by sequential treatment with detergents and deoxyribonuclease. The GBM contains scattered shallow pits representing the sites of immune complex deposits removed in the preparation of the tissue. (×5,000.) (Courtesy of Dr. Stephen M. Bonsib.) |
has certain limitations. First, the staging system provides insight into the evolution of disease but should not imply that all cases necessarily progress from stage I to stage IV. Second, it is often difficult to classify a case of MGN into a discrete stage because multiple stages may overlap, particularly when a fresh generation of deposits develops over an older layer of intramembranous deposits (85,86). Third, the morphologic stages do not correlate well with the level of proteinuria, renal function, or outcome. Fourth, progression from one stage to another can be associated with clinical improvement or worsening of disease. For example, transition from stage II to III to IV may be seen in the setting of resolution of clinical disease, while a patient may progress to advanced renal failure with biopsy findings of only stage II to III. Finally, clinical remission may be associated with no change in the appearance of the deposits, evolution to stage IV, or, less commonly, complete resolution with restoration of a normal glomerular architecture (27,87,88,89,90).
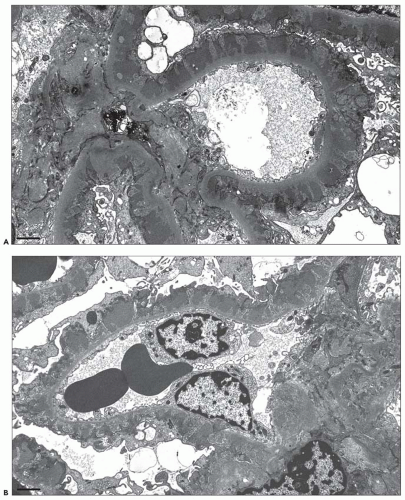 FIGURE 7.22 Electron micrograph of stage II MGN. Images (A) and (B) show the typical findings of stage II MGN, including global subepithelial electron-dense deposits separated by intervening GBM spikes. (A and B: ×6000.) |
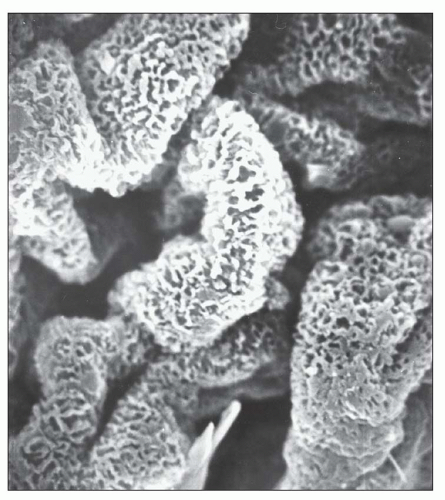 FIGURE 7.23 Scanning electron micrograph of an acellular glomerulus prepared as described in Figure 7.21 from a patient with stage II MGN. The projections of basement membrane material are seen as a complex anastomosing network on the outer surface of the GBM. (×4,000.) (Courtesy of Dr. Stephen M. Bonsib.) |
reports of cases in which light microscopy and immunofluorescence strongly suggest the diagnosis of MGN, but EM reveals deposits that exhibit a fibrillar or microtubular substructure (96,98,99,100,101). Based on their ultrastructural appearance, these cases are best considered as examples of fibrillary glomerulonephritis and immunotactoid glomerulopathy, respectively, and should not be considered as variants of MGN (96,98,99,100,101). Podocyte infolding glomerulopathy is a recently described entity that resembles MGN by light microscopy and may or may not exhibit granular positivity for IgG by immunofluorescence (102,103,104). In contrast to MGN, EM reveals podocyte infolding with microspherical and microtubular structures within the GBM. The majority of cases of protein infolding glomerulopathy have occurred in Japan, and many of the patients have evidence of SLE (102,103,104). Although the morphogenesis of this lesion is unknown, the irregular outer contours of the GBM suggest that podocyte cytoplasmic fragments and cell membranes may become trapped in the course of deposit resorption and matrix remodeling. Finally, organized electron-dense deposits can be seen in membranous LN, and occasional cases of apparently primary MGN have unexplained organized deposits.
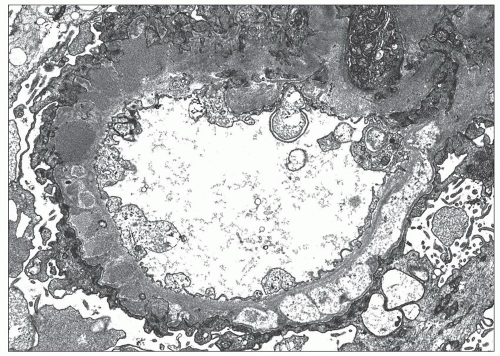 FIGURE 7.24 Electron micrograph of stage III MGN. In this example of stage III MGN, the subepithelial deposits are separated by intervening GBM spikes and accompanied by overlying neomembrane formation. Some of the deposits appear electron lucent consistent with partial resorption. (×6000.) |
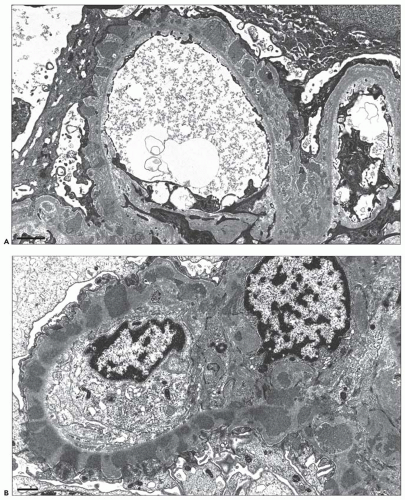 FIGURE 7.25 Electron micrographs of MGN stage II to III. In these two examples of stage II to III MGN, there are global subepithelial deposits, global intervening GBM spikes, and segmental overlying neomembrane formation. Thus, a mixture of stage II and stage III changes is present. (A: ×8000; B: ×10,000.) |
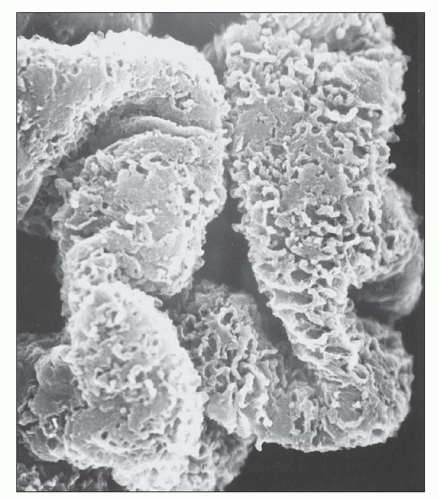 FIGURE 7.26 Scanning electron micrograph of an acellular glomerulus prepared as described in Figure 7.21 from a patient with stage III MGN. The anastomosing ridges of basement membrane material that surround and project above the electron-dense deposits (removed in this material by processing) show areas where they bridge over the deposits and form smooth plaques on the external surface of the GBM. (×5000.) (Courtesy of Dr. Stephen M. Bonsib.) |
ANCA serology was positive by indirect immunofluorescence or ELISA in all cases and was identified as P-ANCA with specificity for myeloperoxidase (MPO) in most cases. The cohort consisted with 8 men and 6 women with a mean age of 59 years. The mean serum creatinine was 4.4 mg/dL, and 12 of 14 patients presented with acute kidney injury (AKI). The mean 24-hour urine protein was 6.5 g/d, all of the patients had evidence of hematuria, and six had extrarenal manifestations of vasculitis (95). The diagnosis of MGN and ANCA-associated GN were established simultaneously in 13 patients, while one had been previously diagnosed with MGN. On pathologic evaluation, the findings of MGN were typically stage I or II and were only segmentally distributed in six cases. Twelve of thirteen patients with available follow-up were treated with steroids and cyclophosphamide; among these patients, 7 had stabilization or improvement in renal function, 1 had worsening renal function, and 4 progressed to ESRD (95).
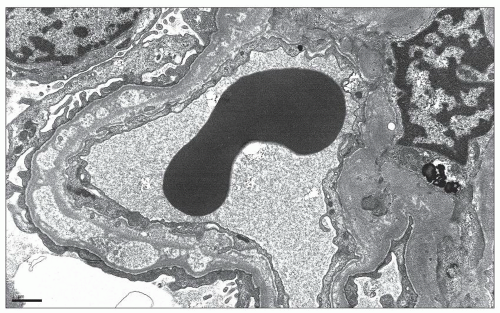 FIGURE 7.27 Electron micrograph of stage IV MGN. In this example of stage IV MGN, there is global remodeling of the glomerular basement membrane which contains intramembranous lucencies consistent with resorbed deposits. (×6000.) |
localized to the subepithelial aspect of the GBM and that the Heymann nephritis model resulted from in situ antigen-antibody complex formation involving an endogenous antigen, rather than circulating immune complexes containing tubular proteins that deposited in the subepithelial region (118,119). In a series of elegant experiments published in 1982, Kerjaschki and Farquhar determined that glycoprotein 330 (gp330) expressed on the proximal tubular brush border was the critical component of FX1a that induced PHN. Specifically, injection of isolated gp330 was able to induce PHN, while injection of FX1a depleted of gp330 did not produce this model (120). As a result, gp330, also referred to as megalin, was identified as the target antigen in Heymann nephritis.
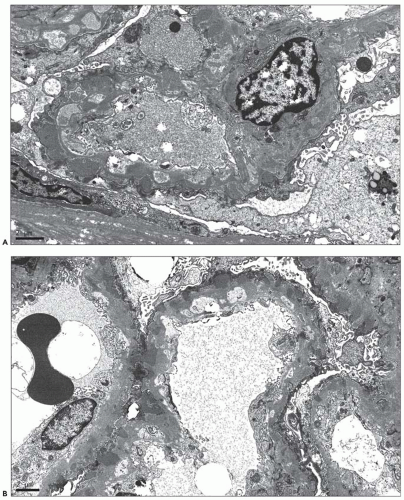 FIGURE 7.28 Heterogeneous deposits in MGN. In these two examples of MGN with heterogeneous deposits, the glomerular basement membrane is expanded by multiple generations of deposits. The deeper layer of intramembranous deposits appears electron lucent consistent with ongoing resorption. The outer layer of newly formed deposits appears electron dense. (A: ×8000; B: ×6000.) |
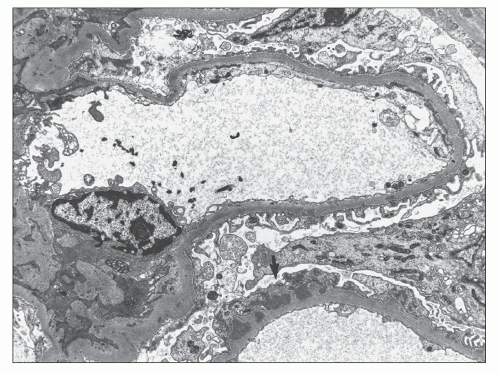 FIGURE 7.29 Electron micrograph of segmental MGN. In segmental MGN, there are segmental subepithelial deposits with intervening GBM spikes (arrow). The adjacent glomerular capillary exhibits only a rare, minute deposit. (×8000.) |
Related posts:
 Development of the Kidney
Development of the Kidney
 Focal Segmental Glomerulosclerosis
Focal Segmental Glomerulosclerosis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


