Clinical Features
Patients with CKD most often present with nonspecific complaints or are asymptomatic and are referred to a nephrologist because of abnormal blood or urinary findings (
10). In symptomatic patients with CKD, often symptoms have been present for months or years (
4). Urinary symptoms suggesting CKD include difficulty with urination, history of urinary tract infections, passage of kidney stones, dysuria, and nocturia.
A common early sign of uremic encephalopathy is sleep disturbance. Typically, patients have difficulty falling asleep, awaken during the night, and again have difficulty falling asleep, with subsequent early morning awakening accompanied by daytime sleeping. Subsequent loss of short-term memory, difficulty concentrating, and episodes of confusion can occur.
Another early sign of CKD is dependent edema that presents first as ankle or periorbital edema. Uremic patients frequently develop congestive heart failure, in which case dependent edema becomes increasingly severe and may progress to anasarca. The skin of uremic patients has a sallow, yellow appearance because it is pigmented with carotene. When blood urea levels are greatly elevated, urea is secreted through the skin where it can dry into a white uremic frost. The patient’s breath has a uriniferous odor because of urea being metabolized to ammonia by mouth bacteria. In terminal stages, uremic patients become delirious and lapse into coma before death. Because of the nearly universal access to RRT, cases of fully developed uremia are rare. Nevertheless, ESRD patients have a high prevalence of nonrenal comorbid disease. With prolonged survival on dialysis, pathologic changes occur in virtually all systems of the body.
Cardiovascular System
CKD is an independent risk factor for CV disease and all-cause mortality. Observational studies indicate that rates of both stroke and myocardial infarction are higher in patients with CKD before the development of ESRD. For example, Go et al. (
11) reported on the risk of all-cause mortality and CV hospitalizations among 1.2 million participants in the Northern California Kaiser Permanente health care system. They found a graded increase in mortality and CV hospitalizations as estimated GFR declined. This association was independent of traditional CV risk factors such as age, blood pressure, diabetes, hypercholesterolemia, and proteinuria and suggests that CKD may be an independent risk factor for coronary heart disease (
11).
CKD markedly increases the risk of CV death from cardiac events and stroke (
2). The mortality risk in CKD patients with CVD is 10- to 30-fold higher than that in normal, age-matched populations. Median survival after an acute myocardial infarction in patients undergoing dialysis is less than 18 months, even in the thrombolytic era. Hypertensive patients with elevated serum creatinine are at higher risk of myocardial infarction and stroke, and diabetic patients with proteinuria are at greater risk for fatal myocardial infarction and stroke (
2). Prevalence of left ventricular hypertrophy (LVH) and congestive heart failure is strikingly elevated in patients with CKD stages 2 through 5, including those undergoing dialysis (
12). Morbidity and mortality for congestive heart failure and coronary heart disease are also excessive in CKD. Moreover, presence of coronary artery disease increases the risk of progression to ESRD in CKD patients. McClellan et al. (
13) observed that patients hospitalized for myocardial infarction or congestive heart failure who also had CKD had a high rate of progression to ESRD in a 12-month period following the cardiovascular event. Current clinical practice guidelines target the treatment of the risk factors of hypertension, hyperlipidemia, and tobacco use not only for the prevention of CV disease but also to retard the progression of CKD (
6).
Cardiomyopathies of End-Stage Renal Disease
The cardiomyopathies of ESRD are classified into dilated or hypertrophic cardiomyopathy (
14). Dilated cardiomyopathy is characterized by enlargement of the chamber of the left ventricle but without an increase in the thickness of the left ventricular free wall or interventricular septum. It is clinically associated with impaired systolic function and a low left ventricular ejection fraction. Dilated cardiomyopathy precedes dialysis in 16% of ESRD patients and develops in another 15% during the course of dialytic therapy (
14). Dilated cardiomyopathy seems to occur most frequently in hypertensive patients who have hyperparathyroidism and high-turnover bone disease. It is suggested that an excessive entry of calcium into myocardial cells limits their capacity to hypertrophy in response to elevated blood pressure. To compensate for the increased workload, the heart is forced to dilate to maintain cardiac output.
Hypertrophic cardiomyopathy is characterized by LVH. Concentric LVH is the most common form of hypertrophic cardiomyopathy in the patient with ESRD and consists of an increased thickness of both the interventricular septum and left ventricular free wall. Eccentric hypertrophy and asymmetric septal hypertrophy do occur but are less common. LVH affects up to 70% of patients during intermediate stages of CKD and approaching 90% of patients with ESRD (
15,
16). Using magnetic resonance imaging, Patel et al. (
17) observed 63.8% prevalence of LVH among 246 HD patients. Independent predictors of LVH were predialysis blood pressure and diastolic LV volume (suggesting volume overload) and the calcium phosphorus product but not anemia. LVH among these patients was associated with LV systolic and diastolic dysfunction. The study suggests the role of preload and afterload in causing LVH and confirms some of the previous studies done with the echocardiograms in HD patients that noted a similar prevalence of LVH (
15,
16,
17). However, LVH has been seen in some patients whose blood pressure appears to be adequately controlled, and the regression of LVH after transplantation suggests that other CKD-specific risk factors are involved (
16). To this end, recent studies have shown that circulating concentrations of FGF23 were markedly elevated in patients with advanced CKD and the elevated FGF23 was independently associated with greater left ventricular mass and greater prevalence of
LVH (
18,
19,
20). FGF23 functions as an endocrine hormone that regulates phosphorus homeostasis through binding to the FGF receptor (FGFR) and Klotho (its coreceptor) in the kidney and parathyroid glands (
21). The authors demonstrated that FGF23 directly induces pathologic hypertrophy of isolated rat cardiomyocytes and that mice develop LVH after intraventricular or intravenous injection of FGF23. In addition, FGFR blocker attenuates the severity of LVH in 5/6 nephrectomized rats without affecting blood pressure (
18). Thus, chronically elevated FGF23 levels may contribute directly to high rates of LVH and mortality in individuals with CKD. Recent studies have also demonstrated the possible role of activation of the mammalian target of rapamycin (mTOR) pathway in LVH. To this end, LVH associated with uremia can be mitigated or even reversed by rapamycin administration in a mouse model of chronic renal failure. LVH also can be modulated by microRNAs, which regulate cardiac growth by degradation of mRNA or inhibition of translocation. The histone acetylation/deacetylation pathway can also modulate the development of LVH (
22). Acetylation relaxes chromatin structure and allows access of transcription factors, whereas deacetylation produces opposite effects.
Atherosclerotic Coronary Artery Disease
Atherosclerotic CV disease is the principal cause of morbidity and mortality in ESRD patients. This largely reflects the prevalence of hypertension and diabetes mellitus in this population and the severity of the atherosclerotic vascular disease that is part of the natural history of these diseases. Nevertheless, severe atherosclerosis can be seen at an unusually young age. In addition, oxidative stress, inflammation, and HDL deficiency and dysfunction contribute to the pathogenesis of accelerated atherosclerosis and CV disease in the ESRD population (
23,
24). In a retrospective analysis of transplantation candidates with no active symptoms of coronary artery disease (CAD), noninvasive imaging revealed abnormalities suggestive of ischemia in 30.7% of patients and coronary angiography showed obstructive disease in 54.1% of the same cohort (
25). Braun et al. (
26) measured cardiac and coronary artery calcification by electronbeam computed tomography to assess coronary artery disease in 49 chronic hemodialysis (HD) patients aged 39 to 74 years. The results were compared with 102 age-matched nondialysis patients with known coronary artery disease who had undergone coronary angiography. A 2.5- to 5-fold higher coronary artery calcification score was found in the dialysis patients, and the scores significantly increased when the patients were reexamined after 1 year.
The high CV mortality in patients with CKD is closely associated with vascular calcification (
27). The severity of calcification in atherosclerotic plaques can be related to plaque size (
28). Although calcification can be found in small plaques, it is most pronounced in larger, complicated plaques having a necrotic lipid core and varying amounts of inflammation (
29). Calcification also occurs along the media of coronary arteries as Mönckeberg medial calcific sclerosis, in which there is little intimal thickening or luminal narrowing (
30). The bone matrix proteins osteopontin, osteocalcin, and osteoblastic differentiation factor (Cbfa1) are expressed in areas of medial and plaque calcification. The presence of these locally produced bone matrix-stimulating factors indicates that metastatic calcification is an active, cell-mediated process similar to osteogenesis rather than a passive deposition of minerals (
31,
32). Moe et al. (
32,
33) and Chen et al. (
31) have suggested that chronic uremia may up-regulate the local deposition of bone matrix proteins and promote atherosclerotic plaque growth.
Hypertension, dyslipidemia, diabetes, and elevated plasma phosphate, homocysteine, and osteoprotegerin levels are risk factors for vascular calcification (
34). Defects in endogenous anticalcification factors may also play a role (
35). In a recent study, Hu et al. showed that CKD is associated with Klotho deficiency, which contributes to soft tissue calcification (
36). By enhancing phosphaturia, preserving glomerular filtration, and directly inhibiting phosphate uptake by vascular smooth muscle, Klotho can mitigate vascular calcification in CKD.
Sudden cardiac death is the single most common cause of death in ESRD patients and accounts for one quarter to one third of all deaths (
2). In an analysis of outcomes of 102 cardiac arrests in HD units in the Seattle area, it was observed that ventricular fibrillation (65%) and pulseless electrical activity (23%) were the two most common reasons of cardiac arrests in HD patients. More than one third of patients died in the dialysis unit, only approximately one quarter were discharged alive from the hospital, and only 15% were alive at 1 year (
37). The possible risk factors for sudden death in dialysis patients include hyper-/hypokalemia and hypervolemia, LVH and heart failure, myocardial fibrosis, QT dispersion, sympathetic overactivity, hyperphosphatemia, and possibly sleep apnea (
38). Unlike nondialysis patients, underlying ischemic heart disease is not an important contributor to sudden cardiac death in the dialysis population (
39). It is unknown whether preventing or treating any of the aforesaid risk factors will reduce the incidence of sudden death in dialysis patients.
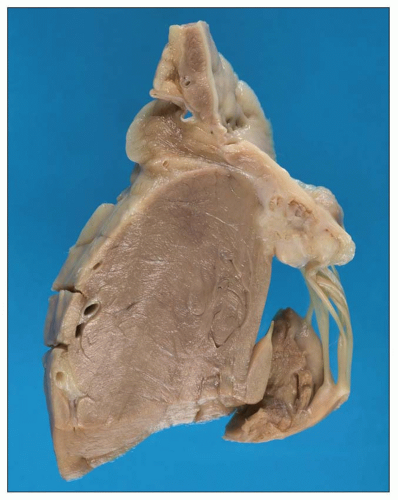
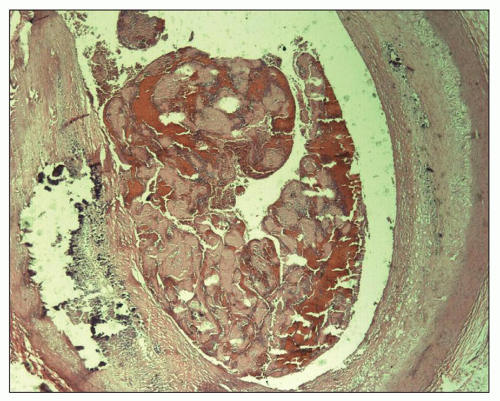
Valvular Calcification
The cardiac valves of ESRD patients are frequently abnormal. Valvular calcification is closely related to coexisting atherosclerotic coronary artery disease, and the extent of valvular calcification has been found to predict CV mortality (
26,
40,
41,
42). The mitral valve is calcified in 10% to 59% and the aortic valve in 28% to 55% of dialysis patients. The calcification most severely affects the annulus (
Figs. 28.1 and
28.2). Although involvement of the aortic valve cusps and mitral leaflets can produce clinically significant stenosis, this is not common, and the relationship between valvular calcification and cardiac death is primarily owing to the close association of valvular calcification with coronary artery disease (
26,
42). Calcification of the mitral valve can cause heart block or other arrhythmias when the calcification of the annulus extends into the adjacent ventricular wall. Oxalosis is sometimes found in the hearts of ESRD patients as a result of the secondary oxalosis of advanced CKD. It can extensively involve the coronary arteries or be deposited in the AV node and conducting bundles.
Calcemic Uremic Arteriopathy
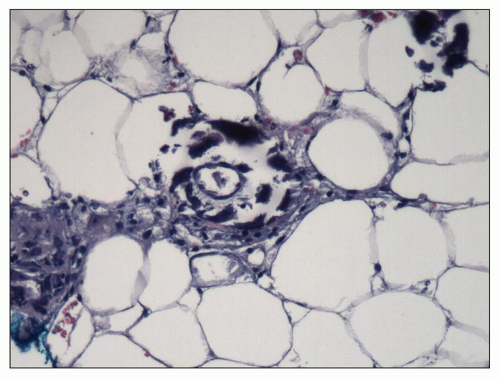
Calcemic uremic arteriopathy (CUA) or calciphylaxis is an uncommon clinical condition that occurs in some patients with long-standing ESRD. Calcification of arterioles in the dermis and subcutaneous tissues produces painful red skin nodules that frequently undergo ischemic necrosis and ulceration (
Fig. 28.3) and is most commonly found on the thighs, buttocks, and abdomen. When the hands or feet are affected, there may be gangrene of the fingers or toes. Involvement of the mesenteric
artery has also been reported. Histologically, the skin shows medial calcification and intimal fibrosis of arterioles and small arteries. The intimal fibrosis may be occlusive or accompanied by thrombosis, and coagulative necrosis is found in the dermis and subcutaneous tissue. Ulceration frequently develops, and a necrotizing panniculitis is sometimes seen that is often complicated by septicemia. Studies have reported incidences ranging from 1% to 4% in chronic HD patients. A case-control study of 67 Japanese patients with CUA identified warfarin therapy, low serum albumin level, elevated plasma glucose level, and increased serum calcium level at the time of diagnosis to be significantly associated with CUA (
43). However, no significant associations were found with female sex, vitamin D analog therapy, serum phosphate level, adjusted calcium-phosphate products, or serum alkaline-phosphatase level. This study suggests that in high-risk patients with poor control of blood sugar and calcium levels, warfarin therapy should be undertaken only with great caution. Treatment of CUA is unsatisfactory. Therapeutic strategies include removing medications thought to contribute to calciphylaxis (calcium-based phosphate binders, active vitamin D analogs, and warfarin) as well as aggressive wound management and antibiotic therapy, supplemented by intensified HD, intravenous sodium thiosulfate, and attempted oxygen therapy including hyperbaric oxygen (
44). Bisphosphonates or cinacalcet may also be helpful in selected patients (
45). There are no randomized control trials, and most of the literature is anecdotal, and results are variable (
46).
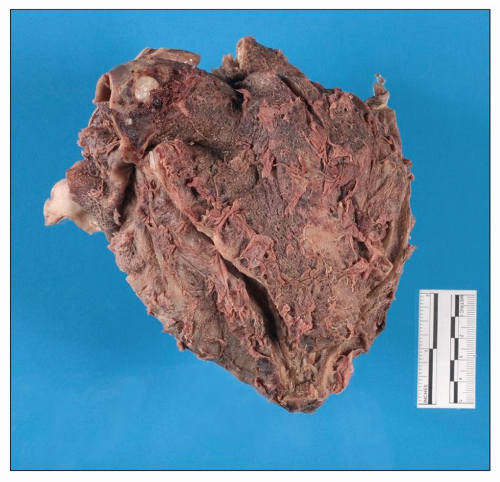
Pericardium
Uremia appears to increase the permeability of small blood vessels and allows a fibrinous exudate to leak across the pericardium leading to
uremic pericarditis (
47,
48). The gross examination of the heart shows easily broken bands of fibrin producing the classical bread and butter appearance (
Fig. 28.4). The histopathology of uremic pericarditis reveals a fibrinous exudate accompanied by sparse inflammatory cells covering the visceral and parietal surfaces of the pericardium.
A fibrinous pericarditis can also be seen in acute renal failure and occasionally in adequately dialyzed patients. When it occurs 1 month or more after beginning dialysis, it is diagnosed as dialysis-associated pericarditis (
47,
48). It is generally held that dialysis-associated pericarditis and uremic
pericarditis represent the same pathologic condition. It is often seen when dialysis patients become infected or when they are under increased metabolic stress, such as following surgery. In most cases, dialysis-associated pericarditis clears after treatment schedules are intensified (
47,
48). In long-standing pericarditis, the fibrinous exudate is organized into fibrous tissue that produces a constrictive pericarditis, which is usually a complication of ESRD, but has also been described with acute renal failure following a single episode of pericarditis. Typically, uremic or dialysis-associated pericarditis is associated with pleuritic chest pain and, if a significant amount of fluid accumulates, with cardiac tamponade (
47,
48). Hemorrhage can occur into the pericardium and is an additional cause of cardiac tamponade in the uremic patient.
Lung
The cardiomyopathy of ESRD frequently decompensates into congestive heart failure as a result of anemia, fluid retention, and hypertension. Congestive heart failure is one of the most common causes of death among ESRD patients. Patients develop pulmonary edema when the passive accumulation of blood within the pulmonary vasculature causes alveolar intracapillary hydrostatic pressure to exceed oncotic pressure. Grossly, the lungs involved by pulmonary edema are heavy, and their sectioned surfaces are covered by frothy, blood-tinged fluid. Microscopically, alveolar capillaries are distended with blood. The perivascular interstitium and interlobular septa are edematous, and lymphatics are dilated. Alveoli become filled with amorphous, slightly eosinophilic edema fluid that can contain a few red blood cells. If patients have been in congestive heart failure for several days, some hyaline membrane formation may be present (
49,
50).
In advanced uremia, proteinaceous fluid sometimes leaks into the alveoli, producing what has been referred to as
uremic pneumonitis, which is a form of pulmonary edema caused by injury to and increased permeability of the alveolar capillaries (
49,
50,
51). It has been suggested that IL-6 is a direct mediator of uremia-induced increase in vascular permeability (
52). Grossly, the lungs have a stiff, rubbery consistency, and the sectioned surfaces have a dark, reddish color. Histologically, the earliest stage reveals a hemorrhagic fibrinous fluid filling the alveoli with hyaline membranes being formed along alveolar walls and alveolar ducts. If the process does not resolve, fibrin in the alveolar fluid and hyaline membranes is organized into whorls of fibrous tissue referred to as
Masson bodies (
50).
When ESRD is complicated by metastatic calcification, it is usually found in the lung. The calcification may be focal or diffuse and tends to affect the upper lobes most severely. Both lungs can be involved. Metastatic pulmonary calcification is usually clinically asymptomatic, and the pattern of deposition may be so delicate that it will not be detected by routine chest radiographs. Nevertheless, if the calcification is extensive, it can lead to respiratory failure. Grossly, the involved areas of lung have a firm but brittle consistency and a fine net-like appearance that exaggerates the outlines of open alveoli. The areas are gray to pale yellow in color and typically contrast sharply with the adjacent normal or congested lung. They are gritty to the touch. Histologically, linear calcifications outline basement membranes within the alveolar septae. Calcifications also are seen along the elastica of arteries, veins, bronchi, and bronchioles and in fibrotic nodules within the lung parenchyma (
50).
Beta-2-Microglobulin Amyloidosis
Beta-2-microglobulin is the light chain of class I HLA molecules, which enters the bloodstream when HLA molecules are degraded and shed from the surfaces of cell membranes. Beta-2-microglobulin accumulates in the plasma of ESRD patients and is deposited as insoluble amyloid fibrils in the articular surfaces of bone, in periarticular connective tissue (
Fig. 28.5), and in
nerve sheaths in as many as 80% of HD patients after 10 years of treatment (
53). It causes carpal tunnel syndrome and a destructive arthropathy of medium- and large-sized joints, mainly of the shoulders and knees (
53,
54). Dialysis-associated amyloid tumors of breast and ovary, nodular skin masses particularly of the buttocks and shoulder area, and nodules of the tongue are additionally described (
55,
56,
57,
58). Beta-2-microglobulin amyloid was found in the hearts of 7 of 18 HD patients who had been dialyzed for more than 10 years (
59). Amyloid was present in the endocardium and myocardium of the left atrium. In the left ventricle, it was localized to the walls of blood vessels and around areas of calcification of the mitral valve. Rarely, amyloid deposits can occur in the intestinal wall (
60,
61).
Beta-2-microglobulin amyloid is found within bone of 19% of HD patients after 10 years of treatment (
62). These bone deposits are responsible for cysts that develop in the carpal bones, femur, and femoral head and possibly for the unusually high prevalence of pathologic femoral neck fractures in HD patients (
Figs. 28.6 and
28.7). Onishi et al. (
62) observed that 62% of patients had femoral neck fractures when periosteal amyloid deposits were found in posterior iliac crest bone biopsies. Only 4% of a control group of HD patients having biopsies who did not show periosteal amyloid had femoral neck fractures.
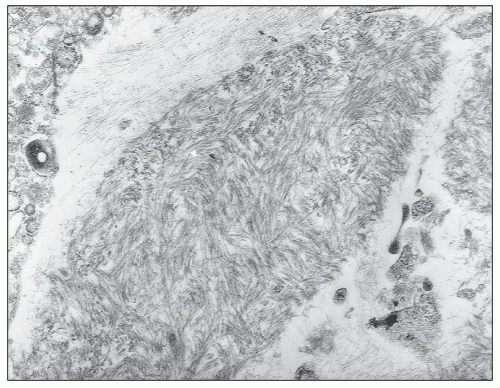
Beta-2-microglobulin amyloid shows Congo red-positive staining and apple-green birefringence under polarized light. By electron microscopy, it is composed of fine fibrils 8 to 10 nm in diameter having a curvilinear structure that is thought to be characteristic of beta-2-microglobulin (
Fig. 28.8) (
62). Since beta-2-microglobulin is cleared to some extent through the peritoneum and PD patients often have more residual renal function, amyloidosis does not seem to be as prevalent with CAPD as it is with HD (
53). The addition of beta-2-microglobulin adsorption columns in tandem with HD reduces the radiolucency of bone cysts in the wrist joints and improves clinical symptoms associated with dialysis-related amyloidosis (
63). A successful renal transplantation often results in marked symptomatic improvement and may arrest the growth of the cysts (
64).
Gastrointestinal Tract and Pancreas
Terminal uremia is accompanied by edema, inflammation, mucosal erosions, and ulcerations of the entire gastrointestinal tract. Gastrointestinal bleeding is a common clinical problem
among dialysis patients and most frequently originates in a hemorrhagic gastritis (
65). Many dialysis patients have a mild gastritis, duodenitis, or peptic ulcer disease in which
Helicobacter pylori can sometimes be found; although, these findings may not be any more common than in the general population (
66).
Colonic bleeding can originate from cecal ulcers, stercoral ulcers, angiodysplasia, diverticulosis, or ischemic colitis (
65,
67). Multiple shallow ulcers attributed to uremia may be found in the cecum and ascending colon. Constipation and fecal impaction, resulting from the use of oral phosphate binders as well as other medications, produce stercoral ulcers in the transverse and rectosigmoid colon. Angiodysplasia consisting of clusters of abnormally dilated mucosal and submucosal blood vessels is found primarily in the cecum and ascending colon. Angiodysplasia is more prevalent in dialysis patients than in the general population, and in some series, it is the most frequent cause of gastrointestinal hemorrhage among ESRD patients (
65,
67,
68). Diverticulosis, diverticulitis, pericolic abscesses, and colonic perforation may be more common in autosomal dominant polycystic kidney disease (ADPKD) than with other categories of renal disease (
69).
The severe atherosclerosis of diabetes and hypertension places ESRD patients at high risk for ischemic colitis and intestinal infarction (
67). HD patients with beta-2-microglobulin amyloidosis may have amyloid deposited in the connective tissue and small blood vessels of the mucosa and submucosa of the GI tract (
60). The gastrointestinal amyloid is usually an incidental finding, but massive involvement of the muscularis propria of the colon has been identified as a cause of bleeding and intestinal infarction (
61).
Systemic inflammation is a constant feature and a major mediator of CV disease and numerous other complications in the ESRD population. Systemic inflammation is associated with and, in part, mediated by endotoxemia, which is invariably present in ESRD patients in the absence of clinically detectable infection (
70). Until recently, little attention had been paid to the role of gastrointestinal tract and its microbial flora in the pathogenesis of the CKD-associated inflammation. However, recent studies have demonstrated marked disintegration of the gastric, small intestinal and colonic epithelial tight junction (
71) and extensive alteration of the composition of colonic bacterial flora in humans and animals with advanced chronic renal failure (
72). More recent in vitro studies have revealed the role of urea and its conversion to ammonia by microbial flora as a major cause of the uremia-induced disintegration of colonic epithelial tight junction and barrier dysfunction (
73). Discovery of the disruption of gastrointestinal epithelial tight junction complex has helped to elucidate the underlying mechanism of endotoxemia and contribution of the intestinal tract to the local and systemic inflammation in ESRD (
70). In addition, via the disruption of the normal symbiotic relationship, the ESRD-associated profound changes in the composition and function of the intestinal microbial flora can contribute to local and systemic inflammation and uremic toxicity. In fact, recent studies have documented the role of colonic bacteria as the primary source of several well-known proinflammatory/prooxidant uremic toxins as well as many asyet unidentified retained compounds in ESRD patients (
74).
Patients dying of uremia may develop an acute pancreatitis in which pancreatic ductules and acini are dilated and contain inspissated eosinophilic material (
75). The dilated acini become disrupted and surrounded by an acute inflammatory reaction. Pancreatitis is also seen in patients who are well maintained on long-term peritoneal and hemodialysis (
76). The pancreas may show focal or generalized fibrosis. Sometimes, fibrosis may be the sequela of episodes of acute pancreatitis, but it has also been related to hyperphosphatemia and to ischemic atrophy secondary to the marked arteriolosclerosis that is often found in the small pancreatic arteries (
75). Nephrogenic or dialysis-associated ascites occurs in a small proportion of HD patients (
77). Some of these patients had been previously treated by peritoneal dialysis (PD), and histologic studies of the peritoneum have shown fibrosis and chronic inflammation.
Hematopoietic System
Anemia is a highly prevalent complication in CKD patients. Contributing factors include erythropoietin (EPO) deficiency, frequently coexistent iron deficiency, and shortened erythrocyte life span. In addition to common causes of iron deficiency that occur in the general population, CKD and more so ESRD patients are at further risk because of impaired gastrointestinal iron absorption, blood loss during the HD, and enhanced incorporation of iron stores into hemoglobin by erythropoietinstimulating agents (ESAs) (
78). A shortened red blood cell life span also contributes to the anemia of CKD. This is reflected in peripheral blood smears that show numerous poikilocytes resulting from extrinsic factors that fragment or metabolically alter the erythrocyte. The anemia of CKD is usually normocytic and normochromic. Since red blood cell production and iron utilization are low, bone marrow examinations of patients with the typical anemia of CKD show normal to increased amounts of iron. Gastrointestinal bleeding and poor dietary intake, however, can lead to iron deficiency and a microcytic hypochromic anemia. Aluminum toxicity causes a microcytic hypochromic anemia in which adequate iron stores are present (
79). Correction of anemia provides relief from many of the debilitating symptoms of ESRD (
80). These include improvement in sleep, cognitive function, and exercise tolerance. The response to EPO is poor in some ESRD patients, and they require high doses of EPO to raise their hemoglobin levels. There are disease differences in EPO requirements. Patients with IgA nephropathy and polycystic kidney disease have lower requirements than other patients, and patients with hypertension appear to need higher doses than diabetics. The differences may be partly explained by underlying inflammatory conditions or infections. Patients with ESRD owing to SLE, vasculitis, and AIDS require relatively high doses, and EPO resistance is more common in African American patients. The hypertrophic cardiomyopathy of ESRD has been observed to regress after EPO therapy, and hemodynamic improvement has been seen with dilated cardiomyopathy (
81). However, administration of high doses of EPO to achieve hemoglobin levels ≥12 g/dL has been shown to increase morbidity and mortality in patients with ESRD and CKD patients not requiring dialysis. These adverse effects are primarily due to the nonerythropoietic actions of high doses of EPO and intravenous iron preparations (
78,
82). Hemorrhage into skin, mucous membranes, and gastrointestinal tract is a common problem in the patient with ESRD. The bleeding tendency is attributed to platelet dysfunction. Platelets are normal in number but appear unable to adequately adhere to damaged blood vessels
and form hemostatic plugs (
83). The ESRD-associated platelet dysfunction is in part due to impaired calcium signaling, which is ameliorated by EPO therapy (
84). In addition, EPO administration increases platelet production and can potentially cause thrombosis in ESRD patients (
85).
Chronic Hepatitis and Hepatic Iron Overload
Hepatitis C virus is the most common cause of chronic liver disease in ESRD. In December 2002, all US chronic HD centers were surveyed regarding selected patient care practices and dialysis-associated diseases. Routine testing for antibody to hepatitis C virus (anti-HCV) was performed on patients at 64% of centers; anti-HCV was found in 7.8% of patients (
86). The prevalence of seropositivity is related to the number of transfusions that the patients have received. Liver biopsies have demonstrated chronic liver disease in 50% to 100% of anti-HCV-positive patients (
87). Martin et al. (
87) found mild or moderate necroinflammatory changes in all of 28 anti-HCV-positive patients who had liver biopsies while being referred for transplantation. Fibrosis was seen in 79%, and 3 of the 28 (11%) had cirrhosis. Hepatitis C infection does not seem to change mortality rates of dialysis patients (
88). The other common liver abnormality in the ESRD population is iron overload, which was due to frequent use of blood transfusion in the preerythropoietin era and is caused by excessive use of intravenous iron preparations in recent years (
89).
Central Nervous System
Central nervous system disorders in ESRD have been defined clinically as uremic encephalopathy, dialysis disequilibrium syndromes, and dialysis dementia (
50,
90). The clinical manifestations of uremic encephalopathy are lethargy, confusion, obtundation, and coma (
90). The signs and symptoms can evolve rapidly in patients with acute renal failure and are less often seen in patients who slowly progress to chronic renal failure. If patients have been severely hypertensive, the brain at autopsy may show fibrinoid necrosis of small arteries and provide evidence of a hypertensive encephalopathy. Otherwise, the brain may reveal only nonspecific changes of cerebral edema, or if patients have been dehydrated, the brain may be dry and reduced in size. The dialysis disequilibrium syndrome is rarely seen in current nephrology practice. The clinical symptoms are caused by elevated intracranial pressure such as nausea, vomiting, tremor, muscle cramps, dizziness, and blurred vision (
90). Dialysis dementia is caused by the chronic toxicity of aluminum absorbed from oral phosphate binders or dialysate water. Aluminum exposure is now closely controlled, and dialysis dementia has virtually disappeared as a clinical syndrome (
90).
Secondary Parathyroid Hyperplasia
In early-stage CKD, plasma fibroblastic growth factor 23 (FGF23), a bone-derived phosphaturic hormone, is increased to maintain neutral phosphate balance through down-regulation of sodium-phosphate cotransporters and suppression of renal production of 1,25(OH)
2D (or calcitriol) by inhibiting 1-alpha-hydroxylase and stimulating 24-hydroxylase (
91). Calcitriol normally acts through the vitamin D receptor to inhibit parathyroid hormone (PTH) transcription and growth of parathyroid cells. Via loss of this inhibitory activity, low serum levels of calcitriol directly contribute to the increased secretion of PTH and promote parathyroid hyperplasia. In addition, low calcitriol reduces the intestinal absorption of calcium and further contributes to secondary parathyroid hyperplasia. In ESRD, dietary phosphate overwhelms the compensatory mechanisms of elevated FGF23, resulting in overt hyperphosphatemia. Hyperphosphatemia together with decreased 1,25(OH)
2D and resulting hypocalcemia contributes to further stimulation of PTH secretion and parathyroid cell proliferation (
91).
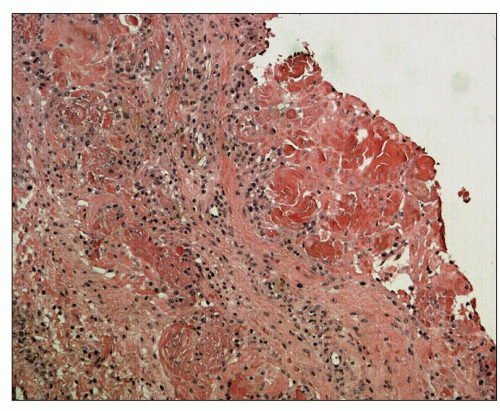
Grossly, all four parathyroid glands and any accessory glands are enlarged in cases of secondary hyperplasia, although there can be considerable variation in the size of each gland. Single-gland weights can range from less than 100 mg to more than 2 g (normal combined gland weight is 50 to 300 mg). The enlargement may be diffuse or nodular. Histologically, the hyperplasia is composed of a mixture of chief cells and oxyphil cells with chief cells usually predominating (
Fig. 28.9). In cases of refractory secondary hyperparathyroidism, clonal chromosomal changes have been detected in the parathyroid glands of 61% of dialysis patients (
92). The most common abnormality was a deletion of 1p that was found in 71% of the glands having chromosomal abnormalities. A deletion of 1p is the most common cytogenetic abnormality found in parathyroid adenomas, and the recurring clonality of the deletion in secondary hyperplasia suggests a neoplastic process and the involvement of a tumor suppressor gene (
92).
CKD-Mineral Bone Disorder and Renal Osteodystrophy
CKD-mineral bone disorder is defined as a systemic condition of mineral and bone metabolism due to CKD manifested by either one or a combination of the following: (a) abnormalities of calcium, phosphorus, PTH, and vitamin D metabolism; (b) abnormalities in bone turnover, mineralization, volume, linear growth, or strength; and (c) vascular or other soft tissue calcification (
93). The term
renal osteodystrophy is used exclusively to define alterations in bone morphology associated with CKD, which can be further assessed by histomorphometry (
93).
It encompasses the following disorders: osteitis fibrosa cystica, osteomalacia, mixed patterns of osteitis fibrosa and osteomalacia, and adynamic bone disease (
94). Osteitis fibrosa cystica is caused by secondary hyperparathyroidism. It is characterized by a high rate of bone formation and resorption and is referred to as
high-turnover bone disease. PTH activates bone remodeling by stimulating marrow stromal cells to proliferate and differentiate into osteoblasts and fibroblasts (
94). PTH also promotes proliferation of osteoclasts either directly or through cytokines and growth factors produced by marrow stromal cells and osteoblasts (
95). The most important of these locally produced factors appear to be macrophage colony-stimulating factor, IL-6, osteoprotegerin, and receptor activator of NF-κB ligand (RANKL).
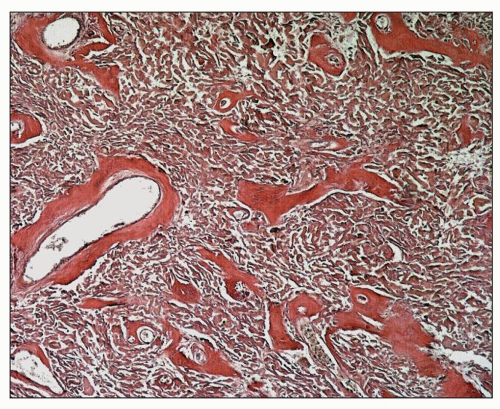
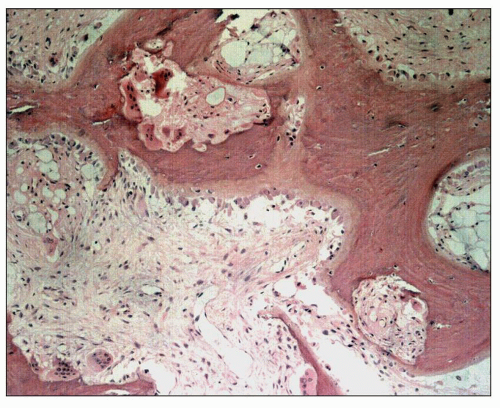
The bones then undergo a continuing process of resorption and deposition that is not under the normal control of local mechanical strain. The cortex of long bones becomes more porous, and the trabecular bone of the medulla becomes thicker as a result of an increase in unmineralized woven bone or osteoid. Histologically, bone biopsies show an increased number of trabeculae composed of lamellar, mineralized bone of irregular shape and thickness lined by increased amounts of osteoid (
94) (
Fig. 28.10). Osteoblasts are prominently clustered along the osteoid. Trabeculae are surrounded by fibrous tissue and are scalloped by osteoclasts within the Howship lacunae (
Fig. 28.11). Unconnected trabecular islands can be seen within the medullary fibrous tissue emphasizing the lack of organization to the remodeling. The bones are susceptible to spontaneous fractures and, in advanced cases, develop medullary cysts and become markedly deformed.
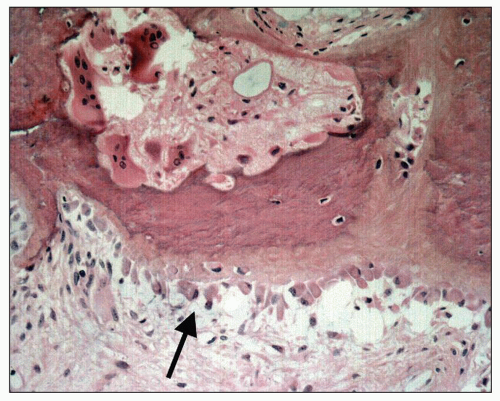
Osteomalacia is a low-turnover bone disease and, in most cases, is the result of aluminum toxicity (
50,
94). Osteomalacia softens the bones that can then become deformed and fracture with mild mechanical stress. Bone biopsies show normal numbers of abnormally thickened trabeculae (
50,
94). The trabeculae consist of a core of irregularly thickened and thinned lamellar bone surrounded by a markedly widened zone of osteoid (
Fig. 28.12). Osteoblasts and osteoclasts are reduced below the number found in normal bone and are sparsely seen in histologic sections. Mixed bone disease shows a background of osteomalacia with foci of osteitis fibrosa in which there is increased osteoblastic and osteoclastic activity (
50,
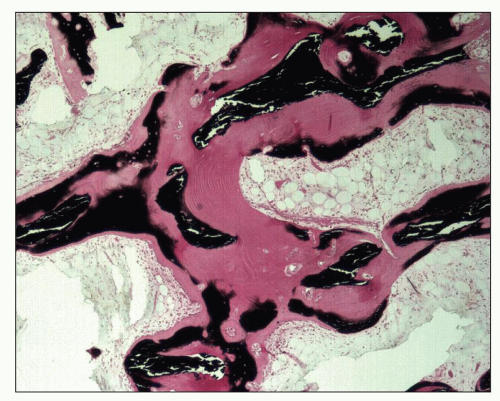
94). Histochemical stains such as alizarin red react stoichiometrically with aluminum. Aluminum bone disease can be diagnosed when a biopsy shows osteomalacia or normal to thinned bone, a low rate of bone turnover, and aluminum histochemical staining over 25% or more of the trabecular surfaces (
96) (
Fig. 28.13).
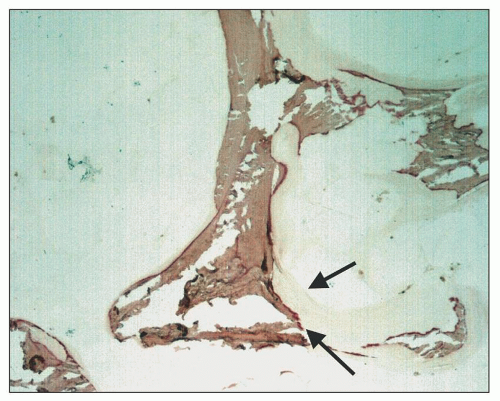
Adynamic bone disease is also a low-turnover state (
50,
94). The underlying mechanism is oversuppression of PTH. It also can be caused by aluminum overload (
97). Patients with adynamic bone disease have bone pain of uncertain cause. In some patients, pain may be the result of microfractures of trabecular bone. In cases of aluminum overload, bone pain can sometimes be rapidly relieved by treatment with deferoxamine. Bone biopsies of adynamic bone disease show normal to decreased numbers of normal-appearing or thinned trabeculae composed of lamellar bone with reduced numbers of osteocytes (
94,
96,
97). The trabeculae show little or no osteoid and few osteoblasts or osteoclasts.
The principal therapeutic approaches for CKD bone and mineral disease include control of hyperphosphatemia/phosphate retention with phosphate binders, administration of vitamin D sterols to suppress PTH, calcimimetic therapy in selected cases of secondary hyperparathyroidism, and parathyroidectomy in refractory cases. All of these measures need to be conducted with attention to the calcium burden since such therapeutic maneuvers can increase the risk of calcium overload with accompanying soft tissue and vascular calcification particularly when using calcium containing phosphorus binders (calcium carbonate and calcium acetate) and vitamin D sterols. Hence, vitamin D administration is not recommended if serum calcium, corrected for albumin, is greater than 9.5 mg/dL or serum phosphorus is more than 4.5 mg/dL (
98). Consequently, novel therapeutic strategies are being developed that utilize calcium-free phosphorus binders (lanthanum carbonate and sevelamer carbonate/chloride) and calcimimetic agents (cinacalcet) to reduce serum phosphorus and suppress PTH without increasing calcium burden (
99).
Hypertension in End-Stage Renal Disease
Ninety percent of patients with CKD experience hypertension (defined as a BP of greater than 130/80 mm Hg) during the course of the disease. Uncontrolled hypertension accelerates the rate of progression regardless of the cause of renal failure. Clinical trials and epidemiologic studies indicate that hypertension is a major risk factor for progressive kidney disease. Evaluation of subjects screened in a multiple risk factor intervention trial who were monitored over a 16-year period showed that (a) higher blood pressure was a strong and independent risk factor for the development of ESRD and (b) the relative risk for ESRD increased with rising systolic blood pressure independent of diastolic blood pressure. In patients with type 2 diabetes mellitus, there is almost a linear relationship between increase in mean arterial blood pressure and yearly decrease in GFR. Analysis of the National Health and Nutrition Evaluation Survey (NHANES) III data suggests that adequate blood pressure control is achieved in only 11% of patients with hypercreatininemia (serum creatinine greater than 1.5 mg/dL) (
100). Analysis of the NHANES IV indicates that only 37% of hypertensive patients with CKD have blood pressure controlled to a level of less than 130/80 mm Hg. Risk factors for uncontrolled hypertension included age older than 65, black race, and presence of albuminuria (
101). In general, older people with hypertension are unaware of their blood pressure elevation, and the majority of those who are aware have poor control rates. CKD prevalence is higher in older age groups, in which systolic hypertension is very common (
2). The importance and potential benefit of blood pressure control in renal outcome cannot be overemphasized.

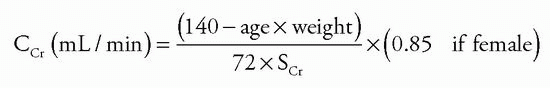
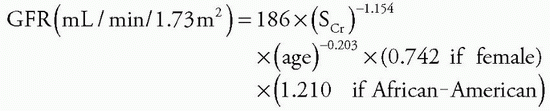
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Glomerular Diseases With Organized Deposits
Glomerular Diseases With Organized Deposits
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis



