Renal Diseases Associated With Plasma Cell Dyscrasias, Amyloidoses, and Waldenström Macroglobulinemia
Guillermo A. Herrera
Maria M. Picken
HISTORICAL PERSPECTIVE
The important historical events more than 150 years ago that brought attention to an association between plasma cell dyscrasias and renal disease deserve recollection. Review of these
historical events allows us to follow chronologically how our understanding of the renal damage associated with dysproteinemias has advanced through the years.
historical events allows us to follow chronologically how our understanding of the renal damage associated with dysproteinemias has advanced through the years.
Thomas Alexander McBean, a tradesman from London, sought medical attention in September 1844 because, while vaulting out of an underground cavern, he felt as if something had snapped within his chest, producing persistent intense pain (1). Dr. William MacIntire, McBean’s attending physician, removed a pint of blood, applied a strengthening plaster to the chest, and recommended abstention from all bodily exertion, resulting in temporary relief and return to his “ordinary avocations.” The improvement did not last long, and further treatment with steel and quinine was performed, with favorable results. However, in the following months, Mr. McBean eventually developed severe weakness, wasting, pallor, hepatic enlargement, pleuritic chest pain, and edema of the face and ankles. These new clinical developments forced a surgeon, whom he consulted, to “take blood from the arm to the amount of 1 pound and to apply leeches and blisters topically.”
Dr. MacIntire observed peculiar abnormalities in his patient’s urine, which was noted to be “opaque, acidic, and of high density with a specific gravity of 1.035” (1). Fifteen months after the initial incident, on October 30, 1845, Dr. Thomas Watson, a leading clinician in London at the time, evaluated Dr. MacIntire’s patient and examined Mr. McBean’s urine, corroborating the previous findings. Seeking help from a well-recognized chemical pathologist, Dr. Henry Bence Jones, was the logical way to proceed. The letter that Dr. Watson sent to Dr. Bence Jones remains an exact description of the urinary abnormalities that are encountered in many patients with renal disease and dysproteinemias. This was the beginning of a saga that deciphered the relationship between a totally unknown blood disorder and the kidney. Dr. Watson stated in this letter:
The tube contains urine of very high specific gravity. When boiled it becomes slightly opaque. On the addition of nitric acid, it effervesces, assumes a reddish hue, and becomes quite clear; but as soon as it cools assumes the consistency and appearance which you see. Heat reliquifies it. What is it? (2)
Dr. Bence Jones took special interest in this specimen, analyzed the urine, and reported his findings. He deduced that the substance responsible for the urine abnormalities was not albumin because it was soluble in acid, and after performing a number of tests, he concluded that it was of a proteinaceous nature and referred to it as an “oxide of albumin, the hydrated deutoxide” (3). He calculated that the patient excreted 67 g/d of this substance. Today, we know this material as Bence Jones (BJ) protein, in recognition of his contribution to our understanding of its nature, in spite of the fact that it was really MacIntire who first discovered the abnormalities in McBean’s urine. Bence Jones was the first to provide a detailed account of McBean’s illness, referring to it as “… a hitherto undescribed disease, essentially malignant in nature … (affecting the) osseous system”; indeed, this is an accurate characterization of a previously unknown disease (3).
Mr. McBean’s condition did not improve. He continued to have excruciating bone pain and developed intractable diarrhea, progressive generalized weakness, and emaciation. He died January 1, 1846, at 46 years of age.
An autopsy performed on Mr. McBean by Alexander Shaw revealed soft, friable ribs, sternum, and vertebrae, and they contained a “gelatiniform substance of blood red color and unctuous feel.” The ribs were “brittle, soft, and easily cut with a knife,” and as described by Dr. MacIntire, they “crumbled under the heel of the scalpel” (4). A diagnosis of mollities et fragilitas ossium, also known at the time as mollities ossium, quite descriptive terms for the disease in question (1,4,5,6), was made. Microscopic sections of the bones were examined by Dr. John Dalrymple, a surgeon at the Royal Ophthalmic Hospital in Moorfields, England, who documented the presence of abnormal cells in detailed drawings he made to illustrate his findings. These cells showed characteristics typical of malignant plasma cells (5), but plasma cells had not even been described at the time. Both Dalrymple and MacIntire believed that the disorder responsible for McBean’s death was essentially a malignant disease of the bone. On his death certificate, the cause of death was “atrophy from albuminuria,” (4) once again alluding to the renal component of this disorder as an essential manifestation of the disease process. The kidneys at autopsy were essentially normal on gross examination. It would take many years of clinicopathologic analysis and research to comprehend the scope of this patient’s disease and to explain the different clinical manifestations.
Although the term multiple myeloma was introduced by von Rustizky in 1873 (7), the disease was rarely recognized until 1889, when Kahler published a case report (8). Kahler recognized that his patient had a similar substance in the urine to that described in McBean’s urine. In 1900, Wright determined that multiple myeloma was a disease of plasma cells (9) when he recognized the similarity of the malignant cellular proliferation in this disease to cells initially described in 1875 by Waldeyer and fully characterized by Ramón y Cajal 15 years later in syphilitic condylomata (10). The association between plasma cells, their secretory products, and nephrotoxicity was not recognized until more than 50 years after McBean’s death, in 1899 (11). Dr. James Ewing, lecturing to medical students in 1932, summarized the available knowledge by stating, “A very peculiar protein (BJ protein), specific of the disease and supposed to be derived from the adsorption of bone” (12). A definitive relationship between BJ proteinuria and the abnormal proteins seen in the serum of patients with myeloma was not demonstrated until 1956 in a study performed by Korngold and Lipari (13). These investigators determined that there were two types of pathologic light chains: κ and λ. Edelman and Gally demonstrated in 1962 that the light chains from the serum and BJ proteins of a myeloma patient were the same (14).
CLARIFICATIONS IN TERMINOLOGY: MULTIPLE MYELOMA AND OTHER MANIFESTATIONS OF DYSPROTEINEMIAS (PLASMA CELL DYSCRASIAS)
There are three clinical entities related to a diagnosis of dysproteinemia: multiple myeloma (often referred to as myeloma), plasma cell dyscrasia (dysproteinemia), and monoclonal gammopathy of unknown significance (MGUS). It should be noted that plasma cell dyscrasia and dysproteinemia are frequently used as generic terms for all of these disorders. Criteria for differentiating these three conditions have been clearly delineated by Durie (15). An understanding of the kinetics associated with plasma cell disorders is important for management and treatment of these patients (16,17).
Myeloma represents the most striking and advanced manifestation of a plasma cell dyscrasia. It is typically associated with lytic (punched out) bone lesions, which are often multiple. There is a monoclonal spike in the serum and/or BJ proteinuria resulting from the production by neoplastic plasma cells of either complete immunoglobulins or fragments of immunoglobulins. Finally, a significant increase in the number of bone marrow plasma cells (usually in the 15% to 20% range), often arranged in sheets with atypical cellular forms, is present. Criteria for clinical diagnosis of myeloma have been proposed (18,19) and approved by the American Society of Hematology.
Myeloma accounts for approximately 1% of all malignancies and 10% of all hematologic neoplasms (20). It is the second most common hematologic malignancy in the United States, with approximately 40,000 individuals suffering from myeloma at any time, and approximately 16,000 new cases are diagnosed every year in the United States (21). At any one time, there are approximately 250,000 patients with myeloma worldwide (21). The incidence of myeloma is approximately 4 in 100,000 individuals; it is higher among blacks than in the general population and more common in males than in females (22,23,24). The disease is most common with advancing age (mean age 65 years), but it is seen in individuals in the fourth and fifth decades of life. It is rare to find it in patients younger than 40 years of age (20), but there are reports of cases in the second decade of life. The incidence of this disease is rising as individuals live longer and survival is increasing. Renal insufficiency is a frequent complication of myeloma and the second most common cause of death after infection in these patients (20,23). Elevated serum creatinine was found in more than 50% of patients with myeloma, at initial examination, in a series of 869 cases described by Kyle (20). Approximately 15% to 20% develop acute renal failure, and a smaller percentage (about 10%) become dialysis dependent (25).
We use the terms plasma cell dyscrasia or dysproteinemia to denote a less than full-blown neoplastic plasma cell disorder. These terms are also often used as generic to refer to any lymphoproliferative or plasma cell disorder associated with production of an abnormal immunoglobulin or light chain. The affected patients often have circulating light chains in the serum or urine detected as a monoclonal (M) spike; they may have clinical manifestations, including renal findings, but the bone marrow is not diagnostic of myeloma. Although there may be a small increase in the number of plasma cells, they are not significantly atypical and are not clustered in sheets. In our experience, in approximately 5% of patients with dysproteinemia, the percentage of plasma cells is within the normal range (<5%). Lytic bone lesions are absent, and clinical manifestations are subtle or nondetectable. Routine bone marrow studies may be incorrectly considered as normal. When ancillary testing is performed (flow cytometry or immunomorphologic evaluations), a clone of plasma cells responsible for the production of the abnormal immunoglobulins is usually found (26). To establish monoclonality in these cases, immunophenotyping can also be performed on cytospin preparations using antibodies that recognize the major Vκ or Vλ subgroups or gene families and those that preferably identify free light chains (FLCs) (27).
The third group of patients with dysproteinemia have an isolated monoclonal M protein peak or gammopathy in the serum, and this condition is diagnosed as an MGUS. The amount of M protein must be lower than 3 g/dL, and there must be fewer than 5% plasma cells in the bone marrow (28,29,30,31,32). Other criteria include the absence or only small amounts of light chains in the urine, absence of lytic bone lesions, and no related anemia, hypercalcemia, or renal failure (28). The individual with an MGUS is otherwise normal. In patients with a diagnosis of MGUS, significant BJ proteinuria, even in the absence of recognizable renal disease, usually precedes clinical and laboratory manifestations of either myeloma or AL amyloidosis, but it may take more than 20 years for a clinical disease to develop (28,29). MGUS is found in approximately 3% of persons older than 70 years of age in Sweden (31). The prevalence of MGUS is higher in older patients, and only 4% of MGUS patients were younger than 40 years in a study by the Mayo Clinic group (28). Some of these patients essentially have “smoldering or indolent myeloma” and, with time, develop full-blown disease. In the Mayo Clinic study, 26% of the patients with MGUS developed multiple myeloma, Waldenström macroglobulinemia, or AL amyloidosis (28). Once a patient with a diagnosis of MGUS develops evidence of organ damage as a consequence of the circulating M protein, the diagnosis of MGUS is no longer tenable. A significant number of MGUS patients eventually develop renal disease. In fact, renal dysfunction is often the first systemic manifestation of progression.
The distinction between myeloma and MGUS based on bone marrow morphology is not reproducible, and it is virtually impossible to unequivocally separate one entity from the other (33). While the percentage of plasma cells is the most predictive feature of myeloma, the cytologic differences are not sharply defined. Interobserver variability is high in the assessment of morphologic atypia of plasma cells, and atypical plasma cells can be seen in patients with MGUS. This emphasizes the importance of identifying renal or other organ involvement in a given patient, because this finding objectively negates a diagnosis of MGUS. Recently, the term monoclonal gammopathy of renal significance has been used to refer to patients with renal manifestations associated with circulating monoclonal proteins and seemingly normal bone marrow evaluations (34).
The fundamental reason for making a distinction between plasma cell dyscrasia and multiple myeloma is because there is far greater consensus regarding the management of myeloma and renal disease compared to patients with renal disease who do not meet criteria for myeloma. The reality is that the pathogenesis for all these disorders is directly related to the overproduction of abnormal monoclonal light or heavy chains by a neoplastic plasma cell clone. The most recent literature stresses the indication for aggressive chemotherapy to eradicate the existing plasma cell clone. In fact, waiting to fulfill the criteria for myeloma before initiating treatment may deny the patient the early intervention that is needed to achieve optimum results.
Synthesis of Immunoglobulin Components by Plasma Cells and Abnormalities in Plasma Cell Dyscrasias
Plasma cells synthesize and secrete specific immunoglobulin molecules often with a minor excess of free κ or λ light chains. The plasma cells synthesize a variety of immunoglobulins, including IgG, IgM, IgD, IgE, and IgA, that can be detected
using serum protein electrophoresis (SPEP). Each immunoglobulin molecule is composed of two identical heavy chains (with molecular weight of approximately 50,000 Da each) and two light chains (molecular weight of approximately 25,000 Da each) linked by variable numbers of disulfide bonds. Both types of light chains consist of a common basic structure composed of a 107- to 111-residue amino-terminal variable (VL) region and a 107-residue carboxyl terminal constant (CL) domain. The VL is the product of two genes, V (variable) and J (joining), that encode the first 95 to 99 amino acids and the remaining 12 amino acids, respectively. The light chain variable region of the germ-line DNA includes multiple V and J sequences. There are approximately 30 Vκ and Vλ germ-line genes that specify proteins on the basis of homology into Vκ 1, 2, 3, and 4 and Vλ 1, 2, 3, 6, and 8 subgroups (35,36,37,38,39,40,41). Variations in the V sequence result from the presence of approximately 30 Vκ and Vλ germ-line genes, with somatic mutations and differences resulting from recombinations of the V and J gene-encoded segments. These variations account for the variability in light chain pathogenicity and the site of pathologic action within the nephron. The carboxyl terminal of each light chain does not vary and is known as the constant C region. Each heavy chain has constant domains (CH1, CH2, and CH3) and a variable domain (VH). There are five types of heavy chains, namely, γ (IgG), α (IgA), µ (IgM), δ (IgD), and ε (IgE). Immunoglobulin G and IgA have variable numbers of disulfide bonds linking the heavy chains to each other and the heavy chains to the light chains. These characterize different isotypes of these Ig molecules, known as IgG1, IgG2, IgG3, and IgG4, as well as IgA1 and IgA2. Immunoglobulin A and IgG2 tend to exist in pairs of units known as “dimers” or may even polymerize to produce larger molecules. Immunoglobulin M exists primarily as a pentamer molecule composed of five Ig units.
using serum protein electrophoresis (SPEP). Each immunoglobulin molecule is composed of two identical heavy chains (with molecular weight of approximately 50,000 Da each) and two light chains (molecular weight of approximately 25,000 Da each) linked by variable numbers of disulfide bonds. Both types of light chains consist of a common basic structure composed of a 107- to 111-residue amino-terminal variable (VL) region and a 107-residue carboxyl terminal constant (CL) domain. The VL is the product of two genes, V (variable) and J (joining), that encode the first 95 to 99 amino acids and the remaining 12 amino acids, respectively. The light chain variable region of the germ-line DNA includes multiple V and J sequences. There are approximately 30 Vκ and Vλ germ-line genes that specify proteins on the basis of homology into Vκ 1, 2, 3, and 4 and Vλ 1, 2, 3, 6, and 8 subgroups (35,36,37,38,39,40,41). Variations in the V sequence result from the presence of approximately 30 Vκ and Vλ germ-line genes, with somatic mutations and differences resulting from recombinations of the V and J gene-encoded segments. These variations account for the variability in light chain pathogenicity and the site of pathologic action within the nephron. The carboxyl terminal of each light chain does not vary and is known as the constant C region. Each heavy chain has constant domains (CH1, CH2, and CH3) and a variable domain (VH). There are five types of heavy chains, namely, γ (IgG), α (IgA), µ (IgM), δ (IgD), and ε (IgE). Immunoglobulin G and IgA have variable numbers of disulfide bonds linking the heavy chains to each other and the heavy chains to the light chains. These characterize different isotypes of these Ig molecules, known as IgG1, IgG2, IgG3, and IgG4, as well as IgA1 and IgA2. Immunoglobulin A and IgG2 tend to exist in pairs of units known as “dimers” or may even polymerize to produce larger molecules. Immunoglobulin M exists primarily as a pentamer molecule composed of five Ig units.
Normal light chains synthesized by the plasma cells maintain a ratio of κ to λ of 2 to 1 in the serum. κ molecules occur predominantly as monomers or noncovalent dimers, with molecular weights of 22,000 and 44,000 Da, respectively, whereas λ molecules typically exist as covalent dimers. The VH and VL comprise the antigen-binding site. The CH2 and CH3 components are involved in effector functions such as binding to immune cells and host tissues and fixing complement. The synthesis of light chains occurs independently from heavy chains, and they combine in the rough endoplasmic reticulum to form the complete immunoglobulin molecule. The fact that light and heavy chains are synthesized independently is the pathogenetic basis for the existence of light chain- and heavy chain-related disorders as specific entities as well as occasional overlap entities (40,41).
In neoplastic plasma cell disorders, there is proliferation of a clone of plasma cells secreting a single type of Ig molecule or subunit that may be identified as a monoclonal peak on SPEP or on urine protein electrophoresis (UPEP) and characterized by immunoelectrophoresis or immunofixation (42). In some cases, only light chains are produced by the neoplastic plasma cells, and they are not generally detectable on SPEP but can be identified in the urine. The demonstration of a monoclonal protein in the serum or urine is important to corroborate a diagnosis of dysproteinemia. In a series of patients with myeloma reported by Kyle (20), a monoclonal protein was demonstrated in 90% of the patients using SPEP. The urine contains light chains in 60% to 80% of myeloma patients as detected by means of UPEP. Urinary light chains (BJ proteins) can also be found in the urine of patients with other B-cell neoplasms with plasmacytic differentiation. While FLCs readily circulate in the body, for heavy chains to be found in the circulation, they need to be released from the endoplasmic reticulum by binding with light chains. This is the reason why heavy chains do not circulate freely in normal individuals. Quantitation of serum FLCs (ratio of free kappa to lambda light chains) is very useful in the diagnosis and follow-up of patients with plasma cell dyscrasias/myeloma (25,43).
During the process of cellular replication and differentiation in the bone marrow, mutations typically take place when mature B lymphocytes are transforming into plasmablasts. The mutated plasmablasts produce a colony of identical mutated plasma cells or what is referred to as a plasma cell clone in a particular bone marrow site. The abnormal plasma cells eventually travel to additional bone marrow locations and other organs, disseminating the pathologic process and producing the various lesions seen in cases of advanced myeloma. Most malignant plasma cell disorders actively produce immunoglobulins, and these are generally composed of one type of light and one type of heavy chain.
In dysproteinemias, the normally controlled production of antibodies is replaced by an inappropriate production of larger amounts of immunoglobulin molecules by the bone marrow. The production of light and heavy chains may be unbalanced, resulting in free light or heavy chains. Imbalance of immunoglobulin production most commonly results in an excess of physicochemically abnormal light chains (39,43).
Furthermore, in dysproteinemic patients, biosynthesis of abnormal light chains, large, polymeric, or fragmented, has been documented in bone marrow cell cultures from patients with monoclonal immunoglobulin deposition diseases (MIDDs) and amyloidosis (40,41). It has become clear that mutations resulting in amino acid substitutions in the light or heavy chain molecules are crucial in determining their pathogenicity or absence thereof, along with the type of renal involvement. In some cases, certain physicochemical characteristics of these immunoglobulin components make them nephrotoxic, and even in cases where the production of these immunoglobulins by plasma cells is small, significant renal damage may occur.
Fewer than 1% of myelomas produce no immunoglobulin molecules (nonsecretory), and approximately 5% to 10% produce only light chains, which may only be detectable in the urine (44). The SPEP in these patients could be normal or shows nonspecific alterations.
The light chains in patients with plasma cell dyscrasia may be larger or smaller than normal, with molecular weights ranging from 12,000 up to 200,000 Da. Normal kappa light chains are monomeric and have a molecular weight of 25,000 Da, while lambda light chains tend to be dimeric with a molecular weight of 50,000 Da (25). Glycosylation of light chains contributes to an increase in their molecular weight. In a small number of myeloma cases (<5%), two different abnormal immunoglobulin molecules or fragments of these molecules are produced, indicating the presence of two distinct clones of neoplastic plasma cells (41). Immunoglobulin G is the most common immunoglobulin produced in myeloma cases (52%), followed by IgA (25%). Myelomas producing IgD, IgE, and IgM together account for fewer than 1% of all cases.
The primary structure of light and heavy chains is mostly responsible for whether a given molecule is pathogenic to the kidney or not, as has been clearly shown in studies with recombinant variable portions of light chains. Not all light chains from patients with plasma cell dyscrasias result in renal damage. Particular amino acid alterations will result in changes in the tertiary conformation of the proteins, leading to either partial or complete unfolding and changes in stability, potentiating aggregation (45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60). The three-dimensional configuration of a given light or heavy chain molecule can be predicted using computer modeling techniques; taking this information into account, the effects of a particular protein can be anticipated (50,52). It is known that λ light chains are preferentially associated with amyloidosis, while κ light chains are most common in light chain deposition disease (LCDD) (54,60,61,62,63). λ Proteins of two Vλ gene families, 6a and 3r, are typically associated with amyloidosis (54), and patients with λ 6 are the most common ones with renal amyloidosis. In LCDD, the majority of patients are κ Vκ 1 or 4 related. The first biochemical characterization of light chain deposits in tissue using extraction techniques was published by Picken et al. in 1989 (57). Changes in the stability and glycosylation of these light chains may also affect their ability to produce renal damage (66). Fanconi syndrome-related acute tubulopathy is almost invariably κ light chain related. In these patients, a specific amino acid substitution in position 30 of the variable portion of the light chain molecule accounts for the failure of complete processing and catabolism of the involved light chain in the proximal tubular lysosomal compartment (65,66). Biosynthetic data from studies of bone marrow plasma cells from patients with myeloma indicate that those light chains, which are heavier than normal, are usually glycosylated, and this alteration can make the light chains more nephrotoxic or change their pattern of nephrotoxicity (64).
Metabolism of Light and Heavy Chains in Normal Individuals and Pathologic Behavior in Patients With Plasma Cell Dyscrasia
Because light chains are low molecular weight proteins, they are freely filtered through the glomeruli and delivered to the proximal tubules. Glomerular clearance of light chains may be affected by a number of factors, including their physicochemical characteristics, size, isoelectric point, hydrophobicity, and state of aggregation. For example, light chain polymers and heavy chains do not cross the filtration barrier. Once the light chains are filtered by the glomerulus, 90% are reabsorbed by the proximal tubules, endocytosed, and catabolized through an endolysosomal process in the apical tubular regions, with their amino acids eventually returning to the circulation (67,68). This process is very efficient in normal individuals, with only a small amount of FLCs found in the urine. The cubilin-megalin receptor located on the brush border of proximal tubular cells working in tandem controls the endocytosis of the light chains (69,70,71). Light chain endocytosis occurs by a very specific, saturable, receptor-mediated process. There are a number of ligands that compete with light chains for brush border binding. The internalized light chains are then transported into vesicles, where the endosomal system catabolizes them. Hydrolytic enzymes present in the endosomes digest the light chains. Some of the process of light chain digestion appears to take place at the brush border itself, before endocytosis. The κ and λ light chain susceptibility to catabolism varies and accounts for the fact that the ratio of κ to λ light chains is reversed in the urine (2:1, λ to κ) (72).
In the setting of a plasma cell dyscrasia, the quantity of light chains in the filtrate may exceed the maximal reabsorptive capacity of the proximal tubular cells. When this occurs, the light chains pass into the distal nephron, where they may precipitate or remain in the tubular filtrate, resulting in light chain (BJ) proteinuria. Light chains precipitate out of solution when the urine is heated to approximately 56°C and redissolve as the temperature rises. As the urine is allowed to cool again, a precipitate forms, followed by dissolution as further cooling occurs. These are the characteristics documented by Dr. Bence Jones in Mr. McBean’s urine (2). Normal patients may excrete small amounts of light chains (up to 50 mg/d), whereas in patients with myeloma, the light chain excretion may increase to 3 to 85 g/d (73,74). There is no apparent specific relationship between some of the characteristics of the monoclonal light chains excreted by individuals (i.e., chemical properties, subtype κ v λ, monomer vs. dimer, anionic vs. cationic) and the presence of pathologic findings demonstrated clinically or experimentally.
Normal light chains are not attracted to the mesangium and do not interact with mesangial cells. In contrast, some physicochemically abnormal light chains from patients with plasma cell dyscrasias interact with purported mesangial receptors and alter mesangial homeostasis.
Also, in patients with myeloma, the concentration of light chains reaching the kidneys is usually much higher, and the inability of the proximal tubules to properly catabolize the abnormal light chains leads to pathologic alterations. In these patients, the light chains commonly circulate as polymers that cannot be properly broken down by the endosomal/lysosomal system in the proximal tubules, enhancing their propensity to produce pathologic alterations. After the glomerular filtration barrier is compromised, as a result of monotypic light chains interacting with the glomerular basement membranes, such polymers may be freely filtered.
While there is a mechanism to deal with the small amounts of light chains that circulate in normal individuals, that is not the case concerning heavy chains, because they cannot be filtered though the glomerulus owing to their high molecular weight. If free heavy chains are released to the circulation, they interact with the capillary endothelium and mesangium, where they engage in pathologic processes. Presumably, the physicochemical characteristics of the particular heavy chains will dictate how they produce pathology. There is no information currently available on how heavy chains are processed by the kidneys, and the knowledge available regarding pathogenesis of heavy chain-related diseases is rather limited at this time. In Waldenström macroglobulinemia, the circulating IgM molecules become entrapped in subendothelial zones and generally do not significantly alter mesangial homeostasis nor produce tubular lesions.
LABORATORY DIAGNOSIS
The identification of a monoclonal protein in the serum and/or urine is important to confirm a diagnosis of dysproteinemia. Immunoelectrophoresis is routinely used to characterize the
monoclonal protein that is detected in serum or urine. SPEP is a good screening test for plasma cell dyscrasia, even though light chain secreting and nonsecreting plasma cell disorders lack a monoclonal spike. In these cases, examination of the urine for BJ proteins is important to make or solidify a diagnosis. The urine must be properly concentrated to detect small amounts of the monoclonal light chains. Immunoelectrophoresis or immunofixation may be necessary to confirm a diagnosis in some instances.
monoclonal protein that is detected in serum or urine. SPEP is a good screening test for plasma cell dyscrasia, even though light chain secreting and nonsecreting plasma cell disorders lack a monoclonal spike. In these cases, examination of the urine for BJ proteins is important to make or solidify a diagnosis. The urine must be properly concentrated to detect small amounts of the monoclonal light chains. Immunoelectrophoresis or immunofixation may be necessary to confirm a diagnosis in some instances.
Immunofixation electrophoresis, a faster technique than immunoelectrophoresis, is the most sensitive and commonly used method available for the detection of monoclonal proteins. It is very helpful in identifying a monoclonal protein associated with a polyclonal increase of light chains, subtle bands associated with faint monoclonal or biclonal proteins, and monoclonal heavy chain fragments in the urine. Clarification of banding patterns noted on electrophoresis gels is possible by direct comparison of results. The superior resolution, simplicity, and enhanced sensitivity of immunofixation make it the diagnostic modality of choice to detect monoclonal gammopathies. One caution with immunofixation is that it requires precise dilution of the antibodies to avoid a prozone effect (42).
High-resolution electrophoresis (thin-layer agarose gels) may be combined with transfer onto nitrocellulose, followed by resolution of bands with monospecific enzyme-tagged antisera or monoclonal antibodies (Western blotting). This procedure is extremely sensitive, is more discriminating than immunofixation, and allows detection of minute amounts of monoclonal light chains (75). The technique can be utilized in selected instances when the monoclonal protein is in very small amounts.
Serum FLCs have become very important in the diagnostic algorithm clinically used to detect and follow plasma cell dyscrasias/myeloma. Serum concentrations of FLCs are dependent upon the balance between production of light chains by plasma cells and renal clearance. The normal serum FLC κ-to-λ ratio is 0.26 to 1.65. A plausible explanation for the inverted κ/λ ratio relates to the kinetics of FLC clearance with kappa molecules normally being monomeric and lambda LCs dimeric (25). If there is an increase of polyclonal plasma cells or renal function impairment, both κ and λ light chains will increase, but the ratio of κ and λ light chains will remain normal. In contrast, a monoclonal increase of either κ or λ light chains by a neoplastic clone of plasma cells will alter the ratio, providing a numerical indicator of clonality. Serum FLC immunoassays provide better sensitivity and precision than electrophoretic tests (25). They are particularly useful in the diagnosis and monitoring of patients with light chain cast nephropathy (myeloma kidney) (76). The high sensitivity of serum FLC immunoassays makes them also very useful in the initial screening for plasma cell dyscrasias.
RENAL INVOLVEMENT IN PLASMA CELL DYSCRASIAS
Renal involvement in dysproteinemia/plasma cell dyscrasias/myeloma is heterogeneous. Approximately 85% of all light chains with plasma cell dyscrasias are nephrotoxic. The morphologic manifestations vary, depending on the renal compartments targeted by the nephrotoxic light or heavy chains. In some instances, more than one renal compartment is affected, and combinations of different patterns of renal damage can be seen in the same patient. The majority of the nephrotoxic light chains (approximately 70%) affect the tubulointerstitial compartment and are referred to as tubulopathic. The other 30% of nephrotoxic light chains preferentially involve the glomerular compartment, producing glomerulopathies (glomerulopathic light chains). The physicochemical characteristics of the involved immunoglobulin molecule appear to be a crucial pathologic determinant. There are also some uncharacterized host factors that may influence the pathologic alterations and the degree of damage. Genetic polymorphism represents an important consideration that has not been studied. In this chapter, the light chain- and heavy chain-associated disorders will be discussed separately, but the reader must understand that on occasions they may be found acting in concert. Each of the diseases has specific clinical manifestations, pathologic findings, pathogenesis, prognosis, and management, and these specific features support viewing them as separate diseases. These diseases include the following:
Light chain (myeloma) cast nephropathy
Proximal tubulopathies, monoclonal light chain mediated
Tubulointerstitial nephritis, monoclonal light chain mediated
Deposition diseases including light chain (L), heavy chain (H), and light and heavy chain (LH) related (LCDD/HCDD/LHCDD)
Amyloidoses including light chain (AL) and heavy chain (AH) related (AL/AH amyloidosis)
Light chain cast nephropathy, proximal tubulopathy, and tubulointerstitial nephritis are part of the spectrum of renal damage produced by tubulopathic light chains. The glomerular and vascular compartments are not typically affected by the tubulopathic light chains. Amyloidosis and the deposition diseases generally exhibit glomerular manifestations, but they are also commonly associated with tubulointerstitial and vascular pathology. In very rare circumstances, alterations in the vasculature (i.e., in AL amyloidosis) may be the predominant (77) or the first morphologic manifestation of renal involvement, preceding pathologic damage to other renal compartments. Combined patterns such as AL amyloidosis and LCDD, LCDD, and light chain cast nephropathy are uncommon (78,79,80) and may alter morphologic expressions of these disorders. For example, in 69% of these cases, in a series of 23 renal biopsies from patients with combined LCDD and light chain cast nephropathy, glomeruli do not display typical nodular glomerulosclerosis and appear essentially normal by light microscopy (81).
Infiltration of the renal parenchyma by neoplastic plasma cells is rare and usually occurs in terminal patients with myeloma (80). Renal insufficiency or failure because of renal parenchymal infiltration is very unusual. Neoplastic aggregates of plasma cells seen in the renal parenchyma (82) may be associated with malignant plasma cells in the urinary sediment (83).
Light Chain (Myeloma) Cast Nephropathy
Historical Perspective
Cast nephropathy was the first renal lesion to be recognized in patients with myeloma. It was well documented by Decastello in 1909 (84), but the first cases had been recorded
in the literature a few years earlier by Ellinger (11). In the early 1920s, Krauss (85) championed the concept of nephrotoxic light chains, but others noted that, at least in some patients, large amounts of BJ proteinuria were not always associated with renal insufficiency. Thannhauser and Krauss (86) hypothesized in 1920 that the tubular casts were concretions of serum proteins and BJ “albumose.” In a comprehensive study by Bell in 1933 addressing renal lesions in myeloma, which included a complete review of the literature, he concluded that “it seems highly probable that casts are the chief cause of renal insufficiency resulting from multiple myeloma” (87). Other series of patients with overt myeloma have shown that light chain cast nephropathy is the most common lesion seen in these patients (88,89,90). This observation has not changed through the years (81,87,88,91).
in the literature a few years earlier by Ellinger (11). In the early 1920s, Krauss (85) championed the concept of nephrotoxic light chains, but others noted that, at least in some patients, large amounts of BJ proteinuria were not always associated with renal insufficiency. Thannhauser and Krauss (86) hypothesized in 1920 that the tubular casts were concretions of serum proteins and BJ “albumose.” In a comprehensive study by Bell in 1933 addressing renal lesions in myeloma, which included a complete review of the literature, he concluded that “it seems highly probable that casts are the chief cause of renal insufficiency resulting from multiple myeloma” (87). Other series of patients with overt myeloma have shown that light chain cast nephropathy is the most common lesion seen in these patients (88,89,90). This observation has not changed through the years (81,87,88,91).
Clinical Presentation and Laboratory Findings
The most typical presentation of cast nephropathy is acute renal functional deterioration or frank renal failure (91,92,93). It remains the most common cause of acute renal failure in patients with myeloma. In some cases, there are identifiable precipitating factors, such as dehydration, hypercalcemia, contrast media, nonsteroidal anti-inflammatory drugs, hyperuricemia, infections, nephrotoxins, or loop diuretics, such as furosemide. Renal biopsy may establish the diagnosis of underlying myeloma, or the patients may already have an established diagnosis of myeloma and are biopsied because of renal insufficiency to determine the renal lesion. After the diagnosis of light chain cast nephropathy, approximately 90% of patients are found to have overt myeloma (89). These patients frequently also have nephrotic range proteinuria, predominantly composed of light chains. Routine urinalysis using a dipstick, which primarily detects albuminuria, commonly fails to pick up light chain proteinuria.
Gross Pathology
There are no specific gross features in kidneys with light chain cast nephropathy (82,88,90). The kidneys may have subcapsular pathology, including granularity and occasional petechiae, but these are likely related to vascular disease (90). The mean weight of the kidneys from patients with myeloma cast nephropathy was 166 g in one autopsy series (90).
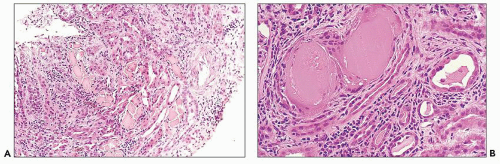 FIGURE 22.1 Light chain cast nephropathy. A: Typical distal nephron casts with brittle consistency, resulting in fracture planes. (H&E, ×350.) B: Multinucleated cell reaction around cast. (H&E, ×500.) |
Light Microscopy
The glomerular and vascular compartments are normal in appearance or show changes related to other preexisting conditions, that is, benign nephrosclerosis. The most striking changes are in the tubulointerstitial compartment when tubular casts are present in the distal nephrons (Fig. 22.1) (91,92,93). In fact, most casts are located in the collecting ducts, and if the medulla is not included in the specimen, the diagnosis may be missed. The typical casts exhibit irregular, angulated, and geometric shapes; fracture planes; and occasionally a lamellated internal appearance, attesting to their protein-rich composition, which imparts to them a firm and often brittle consistency, as they interact with Tamm-Horsfall protein (94,95,96). In some casts, the fragments come together in a jigsaw puzzle-type of arrangement, which is quite peculiar and characteristic (Fig. 22.1A). Casts in the proximal tubules and even in the urinary space are sometimes seen as a result of retrograde filling. An interstitial inflammatory reaction, predominantly with mononuclear inflammatory cells and sometimes eosinophils, often accompanies the tubular casts. Tubulopathic light chains associated with nephron obstruction can elicit an interstitial inflammatory reaction by stimulating cytokines (94,95,96,97,98,99). The interstitial inflammatory process may be as important as the obstructive process (97).
The casts contain predominantly light chains, Tamm-Horsfall protein, and polymorphonuclear cells (99), but they may also include cell debris from tubular damage (94,95,96,98). The epithelial cells in the tubules with casts often appear reactive and, at times, enlarged. Multinucleated cells of purported macrophage origin may also be seen inside tubules surrounding the casts (Fig. 22.1B), and it has been postulated that these cells migrate from the interstitium through the tubular basement membranes into the tubules (100,101,102,103). It also has been proposed that these multinucleated giant cells derive from transdifferentiation of tubular cells to a histiocytic phenotype (104). If the casts break through the tubular basement membranes, then a multinucleated giant cell reaction may be elicited in the adjacent interstitium surrounding the expelled material. These giant cells have also been shown to exhibit a macrophage phenotype. Ultrastructurally, these casts may exhibit fibrils with ultrastructural features of amyloid (105). The casts are generally eosinophilic and generally weakly
periodic acid-Schiff (PAS)-positive; however, there is significant variability in the tinctorial characteristics, as the composition of these casts may be quite variable. In some instances, the tubular casts are PAS-negative, and this may be a helpful diagnostic clue. In rare cases, the casts are composed exclusively of, or contain, crystals (Fig. 22.2). Whereas the morphology of casts may be quite characteristic, there are cases in which the morphology is not pathognomonic. In some of these atypical cases, immunofluorescence may be helpful. However, some cases require careful clinicopathologic correlation for accurate interpretation. The extent of cast formation correlates with the degree of interstitial fibrosis, tubular atrophy, and dropout, and there is also a correlation with renal function in many but not all cases (106).
periodic acid-Schiff (PAS)-positive; however, there is significant variability in the tinctorial characteristics, as the composition of these casts may be quite variable. In some instances, the tubular casts are PAS-negative, and this may be a helpful diagnostic clue. In rare cases, the casts are composed exclusively of, or contain, crystals (Fig. 22.2). Whereas the morphology of casts may be quite characteristic, there are cases in which the morphology is not pathognomonic. In some of these atypical cases, immunofluorescence may be helpful. However, some cases require careful clinicopathologic correlation for accurate interpretation. The extent of cast formation correlates with the degree of interstitial fibrosis, tubular atrophy, and dropout, and there is also a correlation with renal function in many but not all cases (106).
Interestingly, some of the casts are congophilic and, upon polarization, elicit apple green birefringence; they also exhibit thioflavin T and S positivity (105). Certain histochemical and staining properties of renal tubular casts in human multiple myeloma and in “mouse myeloma” are similar to those of amyloid (107).
Immunofluorescence
The glomeruli and vasculature reveal no specific findings. Tamm-Horsfall protein can be demonstrated in the casts, because they form as a result of interactions between this protein and light chains (94,95,96,97,98). Albumin can also be found in the casts. Monotypic (restricted) light chain staining (either κ or λ) of the casts is only seen when the casts have been formed acutely and not when they have remained in place for a prolonged period of time. In a significant number of the cases, there is trapping of the other light chain, and as a consequence, fluorescence staining of similar intensity is noted for both light chains (108). When there is fluorescence for both light chains, the light chain involved in the plasma cell dyscrasia usually predominates, but the degree of one light chain predominating over the other is quite variable and, in some cases, it is difficult to unequivocally determine that there is definitive monoclonality. Silva et al. (103) found that the tubular casts contained the light chain identified in the urine in more than 50% of 40 patients with multiple myeloma.
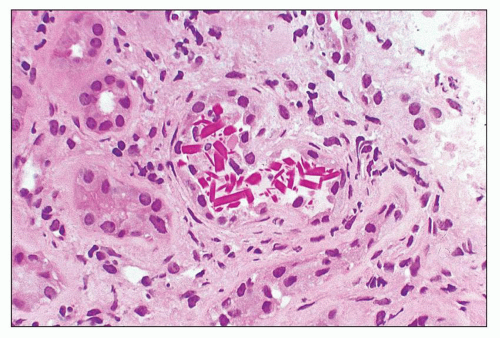 FIGURE 22.2 Light chain cast nephropathy. Distal nephron cast containing crystals. (H&E, ×500.) |
Electron Microscopy
The glomeruli and the vasculature are unremarkable. In most cases, the casts contain abundant fibrillary material admixed with cellular debris. Granular, electron-dense material is seen in many casts, and their specific light chain identity can be substantiated by employing ultrastructural labeling techniques (108,109,110).
In selected cases, the casts are composed of variably sized and shaped crystalline structures. Such casts are fairly specific for light chain cast nephropathy (108,109,110), and the diagnosis can be confirmed by using ultrastructural immunogold labeling to demonstrate monoclonality when immunofluorescence studies fail to make the diagnosis (108,109,110). Immunogold labeling is a far more sensitive and specific technique than immunofluorescence; however, it is not available in most laboratories.
Etiology and Pathogenesis
Casts, in general, form in the distal nephron, and light chain casts are not an exception (85). Local factors combine to optimize cast formation. At this site, Tamm-Horsfall protein, produced by the thick ascending limb of the loop of Henle, is most abundant and provides a perfect nidus for cast formation. The casts form as a result of coaggregation of Tamm-Horsfall protein and light chains (94). The light chains are delivered to the distal portion of the nephron when they exceed the proximal tubule threshold for light chain reabsorption and/or after damage to the proximal tubules impairs reabsorption (95).
It was proposed that a high isoelectric point in the monoclonal light chain predisposed to cast formation (111,112,113,114,115,116), but this theory has not found universal acceptance. Another determinant of cast formation is pH (117). It has been shown that cast-forming monoclonal light chains bind to a common portion of the peptide backbone of Tamm-Horsfall protein, with the carbohydrate moiety in this protein being responsible for facilitating coaggregation. The binding site for Tamm-Horsfall protein on monoclonal light chains is located within the CDR-3 (complementarity-determining region 3) (118,119,120). The secondary structure and key amino acid residues on the CDR-3 of FLCs are important determinants of the molecular interaction with Tamm-Horsfall protein (121). These findings lend support to the current view that the structure of the pathogenic light chain must be such that certain interactions occur. The slower fluid flow in the distal nephron is a contributing factor to effective cast formation.
In 1976, Koss et al. produced an obstructive renal lesion in mice by the intraperitoneal injection of a light chain from a patient with light chain cast nephropathy (122), and Solomon et al. published similar findings in 1991 (111). Clyne et al. proposed in the late 1970s that electrostatic interactions between various proteins involved resulted in precipitation and cast formation (112). Microperfusion of rat tubules with light chains purified from the urine of patients with light chain cast nephropathy has reproduced the distal nephron obstructive lesion in the research laboratory, further attesting to the importance of the physicochemical characteristics of a given light chain in the pathogenesis of the distal nephron lesion (119,120,123,124).
Myeloma casts have been found to be resistant to urinary and macrophage metalloproteinases, making their elimination difficult in some cases (125). The destructive interstitial nephritis that accompanies this lesion has been attributed to
rupture of the basement membranes of tubules with spillage of the cast contents, including Tamm-Horsfall protein, into the interstitium, leading to the release of potent cytokines and other mediators and resulting in potential irreversible interstitial damage (97,126,127), but experimental evidence indicates that direct tubular damage can also activate cytokines leading to recruitment of inflammatory cells (126,128).
rupture of the basement membranes of tubules with spillage of the cast contents, including Tamm-Horsfall protein, into the interstitium, leading to the release of potent cytokines and other mediators and resulting in potential irreversible interstitial damage (97,126,127), but experimental evidence indicates that direct tubular damage can also activate cytokines leading to recruitment of inflammatory cells (126,128).
Differential Diagnosis
The light microscopic appearance of the tubular casts in light chain cast nephropathy is frequently pathognomonic, and there are no other conditions that show similar findings (108). Crystals in casts should create a strong suspicion for a diagnosis of light chain cast nephropathy (108,110). However, when the casts are not classic in appearance, the nephropathologist must carefully evaluate all immunomorphologic data available to make a final determination. When monoclonality cannot be demonstrated by immunofluorescence, the diagnosis of cast nephropathy may be suspected but not confirmed. The clinician must then conduct the necessary studies to confirm the suspicion or rule out this possibility. The differential diagnosis should include nephropathies with cast formation and acute tubulointerstitial nephritis, especially when associated rapid deterioration of renal function. In rare circumstances, light chain protein excretion is increased in unrelated conditions (129). In rifampin-associated light chain proteinuria, a pathologic picture similar to that of myeloma cast nephropathy may occur (130). A similar morphologic picture that has been seen in some patients taking antirejection drugs such as tacrolimus and rapamycin used in combination in cases with delayed graft function can produce intratubular cast formation indistinguishable morphologically by light microscopy from light chain cast nephropathy. However, these casts do not contain monoclonal light chains. It has been suggested that rapamycininduced toxic tubular damage represents an important mechanism in the pathogenesis of this lesion (131).
Treatment, Course of the Disease Process, and Prognosis
The great majority of patients with light chain cast nephropathy have a clearly identifiable plasma cell dyscrasia and meet the criteria for myeloma (approximately 90% of these patients) (89). This is responsible for the alternative diagnosis for this condition: myeloma cast nephropathy. The main therapy of myeloma cast nephropathy is aimed at avoiding the formation of additional casts by reducing the amount of circulating light chains, which is most efficaciously accomplished by treating the plasma cell dyscrasia and facilitating the clearance of existing casts (132). Plasmapheresis, especially in younger patients, has been used to acutely decrease the concentration of circulating light chains, while the chemotherapy decreases plasma cell mass and diminishes light chain secretion (133). This therapeutic strategy is particularly useful in patients with acute renal failure. To facilitate the clearance of existing tubular casts, proper hydration is of utmost importance. Maintenance of a high urine output (aiming at about 3 L/d) is the goal (134). Loop diuretics must not be used, and other agents that promote cast formation or produce renal damage should be avoided. These include radiocontrast agents, nonsteroidal anti-inflammatory drugs, and any nephrotoxic agents. Infections, hypercalcemia, and electrolyte imbalances should be promptly treated. Alkalinization of the urine may facilitate solubility of BJ proteins, but by itself it is of virtually no value.
Colchicine was promoted as an agent that would prevent the formation of new casts as a consequence of its effect on Tamm-Horsfall protein removing its carbohydrate component (94,95,96,135), but the clinical benefit of colchicine treatment remains doubtful. Cysteamine, a reducing agent, may be used to aid in dissolving the existing casts, and vincristine can disrupt casts already in place. Dimethyl sulfoxide has also been used to dissolve casts (136).
While renal function is compromised, many of these patients require dialysis (137,138). Dialysis is also recommended for those with acute-onset renal failure (135,136,138). There is also a role for plasmapheresis to decrease circulating nephrotoxic light chains (97). In addition, recent experimental data in an animal model support a role for small molecule inhibitors of the interaction of Tamm-Horsfall protein and light chains for treating light chain cast nephropathy (121).
Aggressive chemotherapy should be considered seriously and administered to all newly and previously diagnosed patients who are in otherwise relatively good health (132). Alkylating agents and prednisone not only directly act on the proliferating plasma cells but also result in a decrease of proteinuria and directly impact favorably on renal function (132,134,135,136). The treatment of those patients who do not meet minimal criteria for a diagnosis of myeloma is more controversial. Melphalan and prednisone have been used with good initial results in 50% to 60% of these patients (132,136). However, the trend is to be even more aggressive with patients with low tumor burden to effect a cure of the underlying plasma cell dyscrasia.
Dialysis should be instituted early to avoid uremia compounding the usual complications of the disease itself. About 20% of these patients die within the 1st month (138). Renal function improves in about 54% of the patients who present in acute renal failure when the plasma light chain concentration is decreased (132). However, progressive loss of renal function occurs in the majority with time, especially if the myeloma cannot be adequately controlled. The prognosis has not changed significantly in the last 30 years.
The overall median survival for patients with myeloma cast nephropathy and renal failure has been reported to range from 13 months (137,139) to 20 to 30 months, with a 5-year survival rate of 18% to 54% (20,138,139). Renal transplantation is generally not considered a viable therapeutic avenue because of the high risk of recurrence.
Proximal Tubulopathies, Monoclonal Light Chain Mediated (Proximal Light Chain Tubulopathies)
Historical Perspective
Most of the initial publications linking renal damage to myeloma concentrated on cast nephropathy. However, early reports indicated that direct tubular damage by nephrotoxic light chains was an important pathologic mechanism. In 1921, Löhlein first reported crystalline inclusions in proximal tubules in a patient with multiple myeloma (140). The inclusions were also seen in Fanconi syndrome associated with proximal tubulopathy in patients with plasma cell dyscrasias (141). In 1963, Costanza and Smoller (142) and, in 1975, Maldonado et al. (143) suggested that proximal tubular damage was an important pathogenetic mechanism in a subset of patients with myeloma and
renal damage. Clyne et al. (144) injected BJ proteins intraperitoneally in rats and produced intracytoplasmic inclusions in the proximal tubules. However, the glomerular filtration rate did not decline, leading the authors to conclude that although tubular alterations could occur, there was no direct association with renal failure. When these experiments were repeated 5 years later using intravenous infusion of BJ proteins, the same investigators demonstrated that severe reduction in glomerular filtration rate occurred, but in these animals, a significant component of distal tubule cast formation was also noted (112). DeFronzo et al. (145) indicated that the degree of renal failure correlated best with tubular atrophy rather than obstruction and that some patients developed defects in urine-concentrating ability and acidification. Ultimately, however, damage to proximal tubules by some nephrotoxic light chains was clearly demonstrated in experimental nephron microperfusion studies by Smolens et al. (120) and Sanders et al. (123). Some light chains were capable of producing both distal nephron obstruction and proximal tubule damage (123,124). In a clinical study, renal biopsies from patients with myeloma were found to have evidence of proximal tubular damage (146). Pote et al. (126) have further emphasized the role of nephrotoxic light chains in proximal tubular damage and demonstrated experimentally the direct toxic effect of some tubulopathic light chains on proximal tubules. Subsequently, the morphologic spectrum of proximal tubulopathy was demonstrated to include lesions with and without crystalline inclusions, various lysosomal alterations (including “indigestion/constipation”), acute tubular necrosis, and an association with interstitial inflammatory response in some patterns (147,148,149,150). Thus, more recently, the term “light chain proximal tubulopathy” has been proposed to encompass all of the above entities (147,147a,148).
renal damage. Clyne et al. (144) injected BJ proteins intraperitoneally in rats and produced intracytoplasmic inclusions in the proximal tubules. However, the glomerular filtration rate did not decline, leading the authors to conclude that although tubular alterations could occur, there was no direct association with renal failure. When these experiments were repeated 5 years later using intravenous infusion of BJ proteins, the same investigators demonstrated that severe reduction in glomerular filtration rate occurred, but in these animals, a significant component of distal tubule cast formation was also noted (112). DeFronzo et al. (145) indicated that the degree of renal failure correlated best with tubular atrophy rather than obstruction and that some patients developed defects in urine-concentrating ability and acidification. Ultimately, however, damage to proximal tubules by some nephrotoxic light chains was clearly demonstrated in experimental nephron microperfusion studies by Smolens et al. (120) and Sanders et al. (123). Some light chains were capable of producing both distal nephron obstruction and proximal tubule damage (123,124). In a clinical study, renal biopsies from patients with myeloma were found to have evidence of proximal tubular damage (146). Pote et al. (126) have further emphasized the role of nephrotoxic light chains in proximal tubular damage and demonstrated experimentally the direct toxic effect of some tubulopathic light chains on proximal tubules. Subsequently, the morphologic spectrum of proximal tubulopathy was demonstrated to include lesions with and without crystalline inclusions, various lysosomal alterations (including “indigestion/constipation”), acute tubular necrosis, and an association with interstitial inflammatory response in some patterns (147,148,149,150). Thus, more recently, the term “light chain proximal tubulopathy” has been proposed to encompass all of the above entities (147,147a,148).
Clinical Presentation and Laboratory Findings
It is not difficult to conceptualize proximal tubular damage in patients with plasma cell dyscrasias, because light chains are usually metabolized in proximal tubules. The delivery of excessive amounts of physiochemically abnormal light chains to the proximal tubules may lead to overload of the lysosomal system, followed by release of lysosomal enzymes and tubular cell damage. This type of tubular damage may be seen in combination with other renal manifestations in patients with plasma cell dyscrasias, and the clinical manifestations that predominate in those cases may be those related to the other conditions (i.e., those associated with AL amyloidosis or LCDD). When this lesion is found by itself, the clinical presentation may be variable and range from rapidly progressive renal failure, or acute renal failure, to a slowly progressive increase in serum creatinine (76,108,110,142,145,146,147,148,149,150). Some patients with this pattern of renal damage present with proximal tubular dysfunction, including aminoaciduria, phosphaturia, and glucosuria. Other clinical manifestations include subnephrotic range proteinuria, uricosuria, and at times, renal tubular acidosis type II (of proximal tubular origin) (141,142). This is the typical constellation of findings in acquired Fanconi syndrome. Among proximal tubulopathies associated with crystalline inclusions, virtually all cases that have been described have been κ light chain-related, indicating that the composition of the light chains is a crucial determinant of this specific pathologic manifestation (147,148,149). In contrast, proximal tubulopathies not associated with crystals are commonly λ light chain-associated (147,148,149,150). An abnormal clone of plasma cells is detected in approximately 50% of these patients at first presentation (151), and there may be a prolonged interval between the diagnosis of Fanconi syndrome and clinical evidence of myeloma. In one case, the renal abnormalities preceded the demonstration of an underlying plasma cell disorder by 16 years (143).
Similar proximal tubular dysfunction is noted in those cases where the lysosomes present in proximal tubules are filled with monotypic light chains and are unable to release their hydrolytic enzymes (termed “lysosomal indigestion with constipation syndrome”) (147,148,151).
While some degree of clinically insignificant proximal tubular damage may be present in most patients with plasma cell dyscrasias and nephrotoxicity (146), the proximal tubulopathies can be responsible for rapidly or slowly progressive renal failure (110,148). Tubular damage associated with light chain-related Fanconi syndrome occurs in fewer than 5% of patients with renal involvement in plasma cell dyscrasias (93,110).
Gross Pathology
Macroscopic features of kidneys from patients with Fanconi syndrome associated with proximal tubular damage have been documented only rarely. The kidneys have been noted to be enlarged with pale cortical areas (141), findings similar to those seen in association with acute tubular necrosis, regardless of the etiology.
Light Microscopy
Proximal tubular damage by nephrotoxic light chains may lead to crystal formation (tubulopathy with crystalline inclusions) or noncrystalline accumulation of monoclonal light chains in the proximal tubules with variable degrees of tubular damage and lysosomal alterations (108,109,147,148,149,150). In cases with crystal formation, which are frequently associated with clinical Fanconi syndrome, needle-like intracytoplasmic tubular inclusions may be identified with PAS and trichrome stains (146,147,148,149,150,152,153,154,155). Crystalline inclusions may also be seen in the neoplastic plasma cells in the bone marrow (140) and in other cell types (152,153,154,155). There are a few reported cases with Fanconi syndrome containing typical intracytoplasmic crystals in tubular cells coexisting with myeloma cast nephropathy (156). In noncrystalline proximal tubulopathy, three distinct patterns of proximal tubular injury have recently been described: (a) tubular damage with features of acute tubular necrosis, (b) basolateral deposition of the light chain with interstitial inflammatory response, and (c) lysosomal accumulation with enlargement and atypical lysosomal forms (lysosomal indigestion/constipation pattern) (147,147a).
The early changes include vacuolization of the tubular cells followed by apical blebbing with loss of surface microvillous borders, desquamation, and fragmentation (Fig. 22.3A). In some cases, tubular integrity is compromised to the point where only tubular outlines remain (arrows in Fig. 22.3B). Evidence of tubular regeneration with mitotic figures can be seen.
Immunofluorescence
Monoclonal light chains may be detected in the cytoplasm of the tubular cells corresponding to the localization of the light chain in lysosomes (147,147a,148). In other cases, even though staining for both light chains is noted, there is obvious predominance of the pathogenic light chain (110,157,158). This is explained by filtration of the nonpertinent light chain and uptake by the
proximal tubules. The preferential or monotypic staining for a type of light chain should be taken as a clue that the tubulopathy is related to an underlying plasma cell dyscrasia. However, the absence of detectable staining for either of the light chains does not rule out this condition. Sometimes, the abnormal light chains, partially digested in the lysosomes, are not detected by the available antisera. In the Fanconi syndrome-associated cases, the needle-shaped proximal tubular inclusions may fluoresce intensely for κ (extremely rarely for λ) light chains and be very easy to detect (64,65,158). However, in the majority of the cases, fluorescence evaluation does not aid in identifying the cytoplasmic inclusions. Pronase digestion of paraffin-embedded tissues may be of value in detecting the monotypic staining (159).
proximal tubules. The preferential or monotypic staining for a type of light chain should be taken as a clue that the tubulopathy is related to an underlying plasma cell dyscrasia. However, the absence of detectable staining for either of the light chains does not rule out this condition. Sometimes, the abnormal light chains, partially digested in the lysosomes, are not detected by the available antisera. In the Fanconi syndrome-associated cases, the needle-shaped proximal tubular inclusions may fluoresce intensely for κ (extremely rarely for λ) light chains and be very easy to detect (64,65,158). However, in the majority of the cases, fluorescence evaluation does not aid in identifying the cytoplasmic inclusions. Pronase digestion of paraffin-embedded tissues may be of value in detecting the monotypic staining (159).
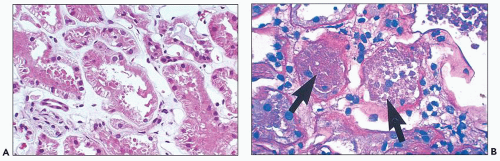 FIGURE 22.3 Proximal tubulopathy, monoclonal light chain related. A: Early mild changes in the proximal tubules, with fragmentation and desquamation of tubular cells. (H&E, ×500.) B: Severe tubulopathy with loss of tubular integrity resulting from cell necrosis (arrows) with loss of nuclei. (H&E, ×750.) |
Electron Microscopy
Proximal tubular damage can be confirmed ultrastructurally. In experimental work and in clinical material, lysosomal proliferation, tubular cell vacuolization and fragmentation, apical cytoplasmic blebs, and segmental loss of microvillous borders are generally present, although in some with variable degrees of severity. The lysosomal system appears overactive, and large and atypical lysosomes are often found (Fig. 22.4) (108,109,110,148,150,160,161). One subset of these patients exhibit proximal tubules packed with large, atypical lysosomes that obscure other organelles (147). In cases associated with Fanconi syndrome, there are needle-shaped, round, or rectangular to rod-like, electron-dense structures in the cytoplasm of the proximal tubular cells. The needle-like inclusion bodies can appear crystalline (Fig. 22.5) or fibrillary (147,147a,162). At high magnification, the crystalline inclusions sometimes exhibit parallel linear arrays. These structures, as well as the large, atypical lysosomes, can also be labeled for the specific light chain using ultrastructural immunogold techniques (Fig. 22.5) (108,109,110,148,158,161).
Etiology and Pathogenesis
The pathogenesis of this type of renal damage is directly related to the inability of the lysosomal system to degrade the nephrotoxic light chains, resulting in overload (“clogging”) of the lysosomes in the proximal tubules with or without crystal formation (123,147,148,160,161). When immunogold labeling is performed, the lysosomes are found to be overfilled with the monotypic light chain that they are unable to properly degrade. In most cases, lysosomal overload causes release of their proteolytic enzymes into the cytosol, leading to cytoplasmic vacuolization, simplification, and even frank necrosis (147). As a consequence, fragmentation, desquamation, and apical blebbing of the proximal tubular cells occur with accompanying segmental or total loss of microvillous borders. Light chains are a ligand for the megalin receptor (70). Silencing megalin and cubilin genes responsible for production of receptor proteins for light chain on the brush border of proximal tubule cells inhibits light-chain endocytosis and ameliorates toxicity (163). Most cases of Fanconi syndrome are associated with the κ1 subgroup, most originating from two germ lines: LCO2 and LCO12 (162). In the case of Fanconi syndrome-associated proximal tubular damage, the partially digested light chains form the fibrillary or crystalline inclusions in the cytoplasm of the proximal tubules. The crystalloid structures have been shown to contain an incomplete monoclonal κ light chain with a truncated NH-terminal fragment corresponding to the variable domain that is necessary for crystallization to occur (64,65,162). These partially digested fragments result from degradation by cathepsin B, and they do not bind Tamm-Horsfall protein except in very exceptional cases. This observation explains why cast nephropathy is so rarely associated with Fanconi syndrome. Elegant experimental work by Sirac et al. (162) has produced a transgenic model of this disorder that closely resembles its human counterpart.
Differential Diagnosis
In proximal tubulopathies without crystalline inclusions, the main differential diagnosis is acute tubular necrosis from other causes. The best way to make an unequivocal diagnosis of light chain-related acute tubulopathy with acute tubular necrosis is by demonstrating monoclonal light chains in association with the lesion in question using immunofluorescence, electron microscopy, immunoelectron microscopy, or a combination of these techniques (108,109,147,147a). Unfortunately, the commercially available antibodies to κ and λ light chains do not always detect the abnormal light chain deposited in the kidneys. Good clinicopathologic correlation may be helpful in solidifying this diagnosis. In the case of light chain-associated Fanconi syndrome, the presence of the characteristic tubular crystalline
cytoplasmic inclusions and the demonstration that these contain monotypic light chains suffice to make a solid diagnosis (147,148,154,155,157,158).
cytoplasmic inclusions and the demonstration that these contain monotypic light chains suffice to make a solid diagnosis (147,148,154,155,157,158).
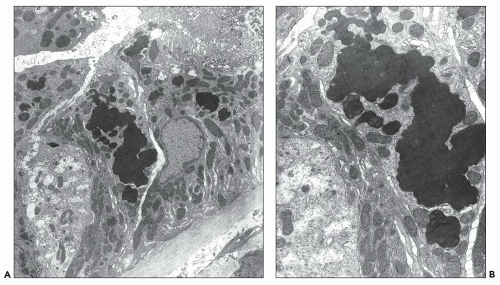 FIGURE 22.4 Proximal tubulopathy without crystalline inclusions, monoclonal light chain mediated. Atypical lysosomes in the proximal tubular cells exposed to tubulopathic light chains with segmental loss of the microvillous border. Transmission electron microscopy. (Uranyl acetate and lead citrate.) (A: ×11,000; B: ×17,500.) |
It is important to consider the possibility of tubular overload being responsible for the staining of light chains in proximal tubular cells. To confirm a diagnosis of light chain proximal tubulopathy, there must be monoclonality for the pertinent light chain and morphologic evidence of tubular damage. Moreover, these findings should be accompanied by clinical evidence of renal dysfunction in order for a diagnosis to be rendered with certainty.
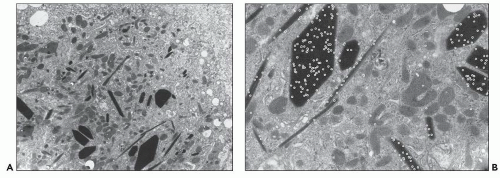 FIGURE 22.5 Acquired light chain-related Fanconi syndrome. A, B: Crystalline cytoplasmic inclusions labeled for κ light chains in the proximal tubular cells. B: Intense gold labeling for κ light chains in cytoplasmic inclusions. Ten-nanometer gold particles have been traced with computer-assisted technology to highlight the labeling. Transmission electron microscopy. (Uranyl acetate and lead citrate.) (A: ×8500; B: ×12,500.) |
Treatment, Course of the Disease Process, and Prognosis
This lesion is often seen in conjunction with other patterns of kidney injury, and the clinical course and prognosis of light chain-associated acute tubular necrosis or tubulopathy as a specific entity have not been critically analyzed. The disease
course and prognosis of the concomitant process tend to prevail in these combined patterns of disease. However, anecdotal cases indicate that, by itself, this lesion is fully reversible if the circulating light chains can be controlled (161). Therefore, aggressive treatment of the underlying plasma cell dyscrasia, together with clinical support while the tubules are regenerating, is the standard of care. Some patients may require temporary dialysis during the acute renal failure episode. It is also important to realize that when combined lesions are present (i.e., LCDD and light chain-associated acute tubular necrosis), the tubulopathy may be the main culprit responsible for the renal failure (110,159,160). If tubular function can be reinstated, renal failure can improve dramatically. This lesion can recur in transplants (148).
course and prognosis of the concomitant process tend to prevail in these combined patterns of disease. However, anecdotal cases indicate that, by itself, this lesion is fully reversible if the circulating light chains can be controlled (161). Therefore, aggressive treatment of the underlying plasma cell dyscrasia, together with clinical support while the tubules are regenerating, is the standard of care. Some patients may require temporary dialysis during the acute renal failure episode. It is also important to realize that when combined lesions are present (i.e., LCDD and light chain-associated acute tubular necrosis), the tubulopathy may be the main culprit responsible for the renal failure (110,159,160). If tubular function can be reinstated, renal failure can improve dramatically. This lesion can recur in transplants (148).
There are variable degrees of tubulopathy, and the mild forms may not be of significant clinical importance. The long-term effects of recurring acute tubular necrosis in the setting of an underlying plasma cell dyscrasia are not known. More careful clinicopathologic studies are needed to clarify the overall importance of this lesion on patients’ prognosis and renal survival.
Because this may be the only renal pathology seen in a biopsy, a definitive diagnosis in a patient with a circulating paraprotein represents objective morphologic evidence of organ damage and should be taken as an indicator that treatment of the plasma cell dyscrasia is warranted.
Tubulointerstitial Nephritis, Monoclonal Light Chain Mediated
This is currently a frequent pattern of renal damage associated with plasma cell dyscrasias (147,147a,164). It mimics acute tubulointerstitial nephritis. It is important to recognize it so that its association with an undiagnosed underlying plasma cell dyscrasia by detecting monoclonal light chain deposition in association with the tubulointerstitial pathology can be established and to rule out other forms of tubulointerstitial nephritis.
Historical Perspective
Two patients were reported in the 1980s with inflammatory tubulointerstitial changes and/or isolated tubular basement membrane monotypic κ light chain deposits. The patients had myeloma with no associated glomerular or vascular light chain deposition, and they were considered atypical LCDD cases (40,165). For many years, it has been recognized that in patients with cast nephropathy, interstitial inflammation may be a significant finding. Some patients with known myeloma and renal insufficiency show no evidence of cast nephropathy or any other forms of plasma cell dyscrasia-associated pathology, but the biopsy shows a patchy (or diffuse) interstitial inflammatory infiltrate associated with tubulitis, providing a clue that such a lesion could be part of the spectrum of plasma cell-associated renal pathology. A series of eight such patients was compiled in 2006 (164) to bring attention to this pattern of light chain-related renal disease, which could be confused with an acute tubular interstitial nephritis unrelated to the plasma cell dyscrasia because of the similarity in histologic findings (166). All but one of these patients and the two previously published cases were κ light chain related, but this lesion may be seen in association with monoclonal λ light chains as well.
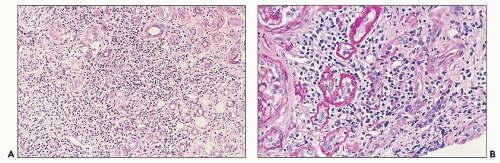 FIGURE 22.6 Tubular interstitial nephritis, monoclonal light chain mediated. A: Intense interstitial inflammation associated with lymphocytes extending through the tubular basement membranes into the tubules. (H&E, ×350.) B: This is better seen on PAS stain. (×500.) |
Clinical Presentation and Laboratory Findings
Among patients with plasma cell dyscrasias and related renal disease, this morphologic pattern accounts for at least 10% of cases (147,147a,164) and is increasingly recognized more often (147a). All the patients with this disease process have presented in acute renal failure, and the cause is either unknown or related to a plasma cell dyscrasia, most often overt myeloma. The patients are usually older than 50 years of age. Serum and urine electrophoresis have shown preferential association with κ light chains. Nonnephrotic range proteinuria may be found. Serum creatinine is quite variable, but it is generally more than 3 mg/dL at presentation (164).
Gross Pathology
The kidneys from two patients exhibiting this lesion in an autopsy series showed normal weight and no specific gross findings (82).
Light Microscopy
The glomerular and vascular compartments are unremarkable. The tubulointerstitial compartment shows an inflammatory process composed predominantly of lymphocytes and plasma cells but also containing variable numbers of eosinophils associated with tubulitis (Fig. 22.6). The inflammatory process can be subtle or intense, focal, or generalized. Tubular damage
can be variable but is frequently marked. There is no tubular cast formation. The absence of casts in multiple sections taken from autopsy kidneys represents substantial evidence that this lesion is clearly separable from light chain cast nephropathy and that it represents a specific pattern of renal damage in patients with plasma cell dyscrasias (164). Because the diagnosis requires the demonstration of monotypic light chains along the tubular basement membranes, it cannot be established by light microscopy alone.
can be variable but is frequently marked. There is no tubular cast formation. The absence of casts in multiple sections taken from autopsy kidneys represents substantial evidence that this lesion is clearly separable from light chain cast nephropathy and that it represents a specific pattern of renal damage in patients with plasma cell dyscrasias (164). Because the diagnosis requires the demonstration of monotypic light chains along the tubular basement membranes, it cannot be established by light microscopy alone.
Immunofluorescence
Linear monotypic light chain staining may be demonstrated outlining the tubular basement membranes in association with the most intense interstitial inflammation. In a small subset of patients, the staining may be focal. There may also be intracytoplasmic staining in proximal tubular cells for the monotypic light chain (147,164). As with acute tubulopathy, there are cases in which staining for both light chains is present, but if the pertinent light chain is more prominently stained, this finding supports the diagnosis.
Immunohistochemistry
In some of these cases, monotypic light chain staining deposition along tubular basement membranes in areas with interstitial inflammation and tubulitis can be clearly demonstrated using immunohistochemistry (164). In these cases, monotypic staining for the pertinent light chain can be striking, whereas there is no staining for the other light chain (Fig. 22.7).
Electron Microscopy
No specific glomerular or vascular changes are identified. Specifically, neither there are light chain deposits nor there is evidence of amyloid deposition. In some cases, there may be focal deposition of light chains, represented by punctate to powdery, electron-dense material along the outer aspect of the tubular basement membranes (147,147a,164). The lysosomal system may be prominent in the proximal tubules. By ultrastructural immunogold technique, distinct labeling for monotypic light chains can be demonstrated along the tubular basement membranes in areas with prominent interstitial inflammation and in tubules exhibiting tubulitis. Both proximal and distal tubules may be labeled, but the findings are usually more striking along distal tubules (108,109,110,164). Highly concentrated monotypic light chains are also noted in proteinaceous material in tubular lumina, which does not organize into well-formed casts (150).
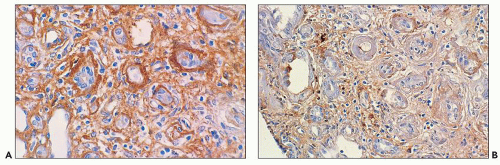 FIGURE 22.7 Tubular interstitial nephritis, monoclonal light chain mediated. Note labeling of tubular basement membranes for κ (A) but not λ (B) light chains in an area with prominent interstitial inflammatory activity. Immunohistochemistry for κ (A) and λ (B) light chains. (Peroxidase antiperoxidase stain, diaminobenzidine as marker, ×500.) |
Etiology and Pathogenesis
The inflammatory interstitial reaction that may occur in these cases is probably induced by the binding of pathogenic light chains to the tubular basement membranes, which alters intrinsic tissue antigens and promotes the release of cytokines, resulting in chemoattraction and activation of interstitial mononuclear inflammatory components (166,167). Light chains may reach the tubular basement membranes by transcytosis after reabsorption or by passive tissue diffusion from peritubular capillaries (164). Conceptually speaking, this pattern of renal disease could be considered as a type of LCDD with pathologic manifestations restricted to the tubulointerstitial compartment (165), but it indeed represents a peculiar, monoclonal light chain-related tubulointerstitial manifestation that warrants consideration here as a separate entity.
Differential Diagnosis
An important differential diagnosis is acute allergic tubulointerstitial nephritis (168,169,170). When eosinophils are present, a hypersensitivity reaction is an important consideration in the differential diagnosis. It is imperative for an accurate diagnosis to demonstrate that monotypic light chains are associated with the tubulointerstitial compartment using ancillary diagnostic techniques. Interestingly, proximal tubulopathy with basolateral monoclonal light chain deposition can also be associated with an interstitial inflammatory response (147,147a). There are no light microscopic findings that allow separation of the light chain-related type of inflammatory tubulointerstitial nephritis from the other types of acute tubulointerstitial nephritis.
Treatment, Course of the Disease Process, and Prognosis
This disease may require steroid therapy, just as needed for any type of tubulointerstitial nephritis (97) and treatment of the plasma cell clone producing the tubulopathic light chains. The use of steroids in this setting is still under clinical investigation,
and no definitive results are available at the present time. There are no specific studies addressing kidney survival or the clinical course of this disease.
and no definitive results are available at the present time. There are no specific studies addressing kidney survival or the clinical course of this disease.
MONOCLONAL IMMUNOGLOBULIN DEPOSITION DISEASES
MIDDs are systemic disorders characterized by deposition of monoclonal immunoglobulins in many organs, but the kidneys are the most commonly involved. In most cases, light chains are the immunoglobulin components that deposit in tissues, giving rise to LCDD. More recently, heavy chain-associated monoclonal deposition disease, also referred to as heavy chain deposition disease (HCDD), has been recognized. Interestingly, the pathologic findings in both light and heavy chain deposition diseases (LHCDDs) are quite similar (89,108,156,171,172). Our understanding of the pathogenesis of light chain-related deposition diseases is more complete than that of heavy chain-related diseases. This is mainly because light chain-associated diseases were described long before their counterparts, and they are more prevalent. Cases of combined LHCDD account for about 10% of all cases (156). The morphologic spectrum of LCDD is extensive, so routine staining of renal biopsies for κ and λ light chains is imperative to identify unusual and early manifestations of these disorders.
Historical Perspective
In 1957, Kobernick and Whiteside showed nonamyloid glomerular abnormalities in patients with myeloma and recognized the similarity of these lesions with diabetic nephropathy (173). About 10 years later, Abrahams et al. (174) reported a myeloma patient with renal disease and deposition of subendothelial material in the glomeruli described as “coarse and more granular than the glomerular basement membrane.” The next year, Rosen et al. (175) reported a similar case, with what he described as glomerular “osmiophilic subendothelial densities.” It was Antonovych et al. (176) in 1974, who first recognized the association of the ultrastructural findings noted above with the deposition of κ light chains. Randall et al. (177) in 1976 published two autopsies from patients with plasma cell dyscrasias and documented the widespread pathologic findings of a disease that he referred to as “systemic LCDD.” HCDD was first described 17 years later by Aucouturier et al. (178), who emphasized that in this disorder, instead of deposition of light chain immunoglobulin components, there were deposits composed of monotypic heavy chains. In 1985, Jacquot et al. (78) were the first to describe three cases of LCDD associated with light chain (AL) amyloidosis.
Light Chain Deposition Disease
Clinical Presentation and Laboratory Findings
There is no significant sex predilection for this disease, but males predominate slightly in most series (7:5). The average age of patients with LCDD with renal manifestations is 55 to 60 years. At the time of diagnosis, acute renal failure is present in 30%, and a similar percentage is dialysis dependent. Most (more than 90%) present with proteinuria, and the average protein excretion per 24 hours is in the nephrotic range (generally 4 to 5 g/d) in 53% of LCDD patients. Interestingly, full-blown nephrotic syndrome is noted in a minority of these cases (156). Seventy-eight percent of LCDD patients present with hypertension, 52% have hematuria, and varying degrees of renal insufficiency are detected in 95% of these patients (155). In selected LCDD cases, tubular dysfunction is the predominant clinical manifestation (179,180). As the clinico-pathologic heterogenicity of this disease is better appreciated, patients are diagnosed earlier, and the manifestations of renal insufficiency tend to be less pronounced (179,180,181,182,183,184).
Rapid deterioration of renal function occurs as the disease advances, if not treated aggressively. Renal biopsy preceded any other clinical signs or evidence of dysproteinemia in 70% of the cases with pure LCDD (156,172). Classic features of underlying myeloma are present in more than half of these patients, but a significant number of patients with LCDD have either a normal bone marrow biopsy and aspirate or rather unimpressive plasmacytosis at presentation (108,110). Careful evaluation of the plasma cells in the latter case using either immunohistochemical techniques or flow cytometry can identify a monoclonal population of plasma cells, albeit a small clone in many cases. Biosynthetic studies of the plasma cells often demonstrate paraprotein production, even when serum and urine electrophoresis fail to show any abnormalities (40). In autopsy of patients who died with a clinical diagnosis of myeloma, approximately 3% to 5% exhibited LCDD (78,84). This figure is conservative, as the less characteristic cases of LCDD are not diagnosed because immunofluorescence and electron microscopy of the kidneys were not part of the evaluation of these autopsies. The most commonly involved light chains in these cases are κ, and subtypes IV and I predominate (185). The ratio of κ to λ is 9:1 in LCDD (156).
Bone marrow biopsy and aspirate revealed sufficient criteria to diagnose myeloma in 35% of LCDD cases, osteolytic lesions in 13%, and hypogammaglobulinemia in 33%. Of the patients with LCDD at the time of clinical presentation and diagnosis, 39% carried a diagnosis of MGUS (186). Up to 30% of patients with MIDD have no detectable monoclonal proteins in urine or serum. Even with use of the most sensitive techniques available, the percentage of patients falling into this category remained at 15% to 20% (186).
When LCDD coexisted with light chain cast nephropathy, 82% of these patients presented with acute renal failure, and 64% required dialysis (55,89). Renal biopsy preceded any clinical evidence of dysproteinemia in 64% of the patients with combined LCDD and cast nephropathy. Cases of combined cast nephropathy and LCDD are mainly associated with κ light chains. LCDD can also occur in combination with light chain-associated (AL) amyloidosis (75,76,77) and HCDD, in which case it is called LHCDD (156). The paraprotein composition of LHCDD is typically IgG-κ or IgG-λ (156).
Gross Pathology
There are only two autopsy studies in patients with myeloma performed after LCDD was described in 1976. Three patients with LCDD were noted in the series by Ivanyi et al. (88,187), and the mean weight of the kidneys was 271 g. Herrera et al. (82) reported the autopsy findings in 77 patients with myeloma. These cases comprised approximately 10% of all cases with myeloma that died during the period of time this study covered at the institution. There were two cases with LCDD, both with small kidneys with average weight of 130 g and with evidence of surface scarring attributed to coexistent vascular disease.
Light Microscopy
The most characteristic finding in patients with LCDD is nodular glomerulopathy (Fig. 22.8) that mimics the pattern of nodular glomerulosclerosis in diabetic nephropathy (108,188,189,190,191,192,193,194,195). Capsular drop and hyaline cap lesions are not present in LCDD, and the absence of these features helps in the differential diagnosis from diabetic nephropathy. In addition, the mesangial nodules in LCDD are more evenly distributed, although there is significant variation depending on the stage of the disease process (60,61,184,189,190,194,195), whereas the nodules in diabetic nephropathy tend to be asymmetric. The mesangial nodules are argyrophilic and composed of extracellular matrix proteins admixed with monotypic light chains (196,197), and the principal matrix protein deposited is tenascin (197). In silver methenamine-stained sections, there may be lamellation of the peripheral portions of the mesangial nodules. Mesangial hypercellularity accompanies the increase in extracellular matrix in some of the cases. The peripheral capillary walls are variably thickened, and the capillary wall alterations are uneven from one glomerulus to the other and as well as within the same glomerulus. The thickened walls are a consequence of the subendothelial deposition of light chains (110,156). There are a number of glomerular morphologic patterns, including mesangial, membranoproliferative, and crescentic (194,195,196,197,198,199,200,201), that precede the nodular glomerulopathy. Progression from one of these “early” patterns to a classic nodular glomerulosclerosis has been shown to occur over time using repeat kidney biopsies. A recognized “atypical” variant of LCDD shows peculiar light microscopic features and ultrastructural findings (202) simulating an immune complex-mediated disease. The light chain deposits, regardless of whether they are in renal or extrarenal sites, are always Congo red negative.
In patients with early LCDD, the glomeruli at times appear essentially unremarkable, and these cases may be incorrectly diagnosed as minimal change glomerulopathy if immunofluorescence staining for light chains is not performed (81,108,109,157,172). Recognition of this LCDD pattern is very challenging (109,110,172) and requires careful immunomorphologic evaluation and/or immunoelectron microscopy (108,157,172) to confirm the association of monoclonal light chain deposits with the capillary walls.
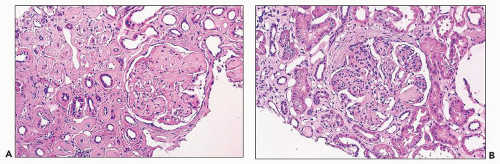 FIGURE 22.8 Light chain deposition disease. Nodular glomerulopathy characteristic of LCDD is evident in (A). Note variation in cellularity, with hypercellular nodules present in (B). (H&E, ×500.) |
The extraglomerular changes may also be quite impressive. The tubular basement membranes may be thickened and tortuous, as a result of deposition of light chains, generally on the outer aspect of the tubular basement membranes (Figs. 22.8A and 22.9). In our experience, thickening of vessel walls by light chain deposits is seen in approximately 40% of LCDD cases. In half of LCDD patients with vascular changes, concentric thickening of the small and medium arteries accompanied by focal light chain deposits (172) creates a striking hyperplastic vasculopathy. The interstitial deposits are sometimes visible by light microscopy, where they form PAS-positive aggregates and are rarely associated with giant cell reaction. Light chain deposits can be found in many organs, including the lung, liver, small and large intestine, thyroid, prostate, pancreas, rectum, skin, spleen, and choroid plexus, among others (57,177,182,183,203).
Immunofluorescence
Deposition of monoclonal light chains can be seen along the peripheral capillary walls in the glomeruli, alongside the tubular basement membranes, and in the vessel walls (Fig. 22.10); the staining is usually diffusely linear and regular but, in some cases, can be interrupted or subtle (55,57,108,110,159,183). The pattern and intensity of staining depend on the amount and distribution of light chain deposits. In most cases of LCDD, the pathogenic light chain is of kappa isotype. No staining is noted for the other light chain, defining the monotypic nature of the labeling pattern. In most cases, there is also granular monotypic light chain mesangial staining in the glomeruli, but mesangial staining is rare without accompanying capillary wall staining. In a subset of these patients, there is also focal granular staining for the pertinent light chain in the interstitium proper. Staining for immunoglobulin heavy chains (IgG, IgM, and IgA) as well as complement components C3 and C1q are typically negative. Rare atypical cases of LCDD have been associated with positivity for complement components (204). In some cases, there is a discrepancy between the fluorescence staining and the demonstration of light chains by electron microscopy. In fact, there are a few reported cases of combined LCDD and light chain cast nephropathy, in which the monotypic light chain deposition has been demonstrated by immunofluorescence and no corresponding deposits were found ultrastructurally (89,156). Such
cases often accompany light chain cast nephropathy and are referred to as “LCDD by immunofluorescence only.”
cases often accompany light chain cast nephropathy and are referred to as “LCDD by immunofluorescence only.”
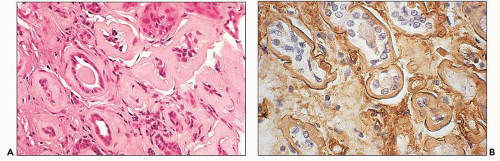 FIGURE 22.9 Light chain deposition disease. A: Thickened tubular basement membranes. (H&E, ×500.) B: κ Light chain deposition on the outer aspect of the tubular basement membranes. (Immunohistochemistry for κ light chain, peroxidase antiperoxidase stain, diaminobenzidine as marker, ×500.) |
Generic antibodies to κ and λ light chains cannot detect all pathologic light chains deposited in the kidney in patients with LCDD. Only when a specific antibody is raised against the pathologic light chain can it be guaranteed that the light chain will be detected. Also, in patients with concomitant diabetes mellitus and LCDD, the abnormal light chains are frequently glycosylated to a degree that impairs their detection. The usual diffuse linear staining seen along peripheral capillary walls in diabetic patients may make it difficult to identify a monoclonal light chain. Immunogold electron microscopy is successful in labeling the abnormal light chains even when generic antibodies are used, attesting to the increased sensitivity of this technique (157). Immunofluorescence techniques have detected deposits in virtually every organ (183,203).
Electron Microscopy
Light chain deposition manifested by flocculent to granular to powdery electron-dense material can be seen in all renal compartments (Figs. 22.11 and 22.12), especially in cases with nodular glomerulosclerosis (advanced lesion). Visualization of light chain deposits may be challenging because the deposits may blend with the mesangial matrix and glomerular/tubular basement membranes. In the glomerular capillary walls, the deposits tend to form thin band-like powdery deposits along the lamina rara interna. Larger deposits may pool in the subendothelial regions. In rare cases, the electron-dense light chain material infiltrates the lamina densa, mimicking dense-deposit disease (108,109,205). At low magnification, the light chain deposits may simulate immune complexes (202). Powdery electron-dense deposits are also generally present along the outer aspect of the tubular basement membranes, along the Bowman capsule, and in the interstitium proper (Fig. 22.12A) (206,207). There may also be distinct deposition of light chains in the vasculature, where they tend to deposit in the intima and the basement membrane cuffs that surround individual medial myocytes (Fig. 22.12B). The light chain deposits may be subtle when the amount of light chain deposits is limited (108,109), and they may blend with the surrounding structures. In combined MIDD and cast nephropathy cases, the light microscopic alterations in the glomeruli and other
compartments may be subtle and easily missed, but combining the data obtained from immunofluorescence and electron microscopic evaluation should suffice to make the correct diagnosis (Fig. 22.13) (81,89,156).
compartments may be subtle and easily missed, but combining the data obtained from immunofluorescence and electron microscopic evaluation should suffice to make the correct diagnosis (Fig. 22.13) (81,89,156).
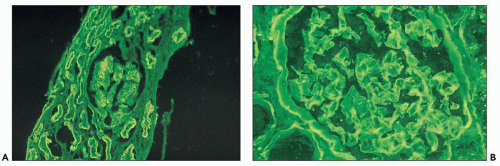 FIGURE 22.10 Light chain deposition disease. A: Low-power view showing linear staining along the tubular basement membranes and mesangial staining in the glomeruli for κ light chains. (Fluorescein, ×150.) B: Linear peripheral glomerular capillary wall staining and staining along the tubular basement membranes and Bowman capsule. Direct immunofluorescence for κ light chain. (Fluorescein, ×500.) |
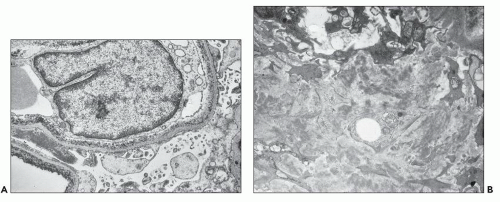 FIGURE 22.11 Light chain deposition disease. A: Continuous, punctate, subendothelial, electron-dense material (light chains) outlines the peripheral glomerular capillary wall. (Uranyl acetate and lead citrate, ×8500.) B: Mesangial light chain deposition. Transmission electron microscopy. (Uranyl acetate and lead citrate, ×9500.) |
Etiology and Pathogenesis
Conclusive evidence that the abnormal light chain protein is primarily responsible for light chain-associated disease has been provided from an in vivo model using mice injected with human BJ proteins from patients with LCDD. Solomon et al. (62) demonstrated nodular and diffuse light chain precipitates in the tubular basement membranes, mesangium, and vessel walls, recapitulating the key findings in the kidneys of these patients. Khamlichi et al. (49) confirmed these results.
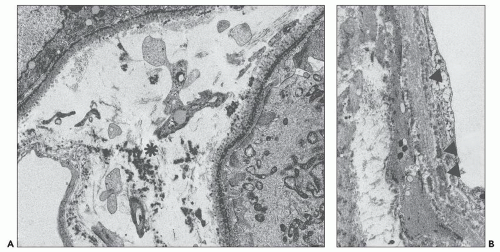 FIGURE 22.12 Light chain deposition disease. Punctate, electron-dense material along the tubular basement membranes and in the interstitium proper (asterisk in A) and along the vessel wall (arrowheads in B). Transmission electron microscopy. (Uranyl acetate and lead citrate.) (A: ×7500; B: ×9500.) |
Since then, the pathogenesis of LCDD has been scrutinized using renal biopsy material and an in vitro mesangial cell culture model. Studies on renal biopsies demonstrating plateletderived growth factor-β (PDGF-β) and transforming growth factor-β (TGF-β) in the mesangium of patients with LCDD in various morphologic stages suggested a role for these two growth factors in the pathogenesis of this disorder (208). The in vitro model confirmed the findings in the renal specimens
and showed that interactions between mesangial cells and light chains from these patients were crucial in the pathogenesis. Rat and human mesangial cells grown on cover slips and in a matrix with composition similar to that of an altered mesangial “milieu” and incubated with light chains purified from the urine of patients with LCDD recapitulated the biopsy findings (209,210,211,212,213). Human mesangial cells incubated with light chains purified from the urine of patients with LCDD transform from their usual smooth muscle to a myofibroblastic phenotype (214), endowing them with the necessary machinery to engage in active synthesis of extracellular matrix proteins. The initial events were activation of PDGF-β, leading to cellular proliferation through c-fos activation, and later TGF-β activation, resulting in matrix deposition (203,211,212,213). The in vitro model clearly demonstrated the crucial role of TGF-β in the production of extracellular matrix proteins by mesangial cells in LCDD (213). Tenascin is the main extracellular matrix protein in the nodules formed in the in vitro model as well as in the mesangial nodules in nodular glomerulosclerosis in LCDD (197,210,211) by transformed mesangial cells with a myofibroblastic phenotype (214). Tenascin is mainly degraded by matrix metalloproteinase-7 (MMP-7), with minor contributions by MMP-1 and MMP-3 (215). MMP-7 secretion by mesangial cells is impaired in LCDD, making tenascin degradation virtually impossible (216). The specific mechanism responsible for this has not yet been elucidated. The interactions between the light chains responsible for LCDD and mesangial cells occur through a receptor-mediated mechanism (217) (Fig. 22.14).
and showed that interactions between mesangial cells and light chains from these patients were crucial in the pathogenesis. Rat and human mesangial cells grown on cover slips and in a matrix with composition similar to that of an altered mesangial “milieu” and incubated with light chains purified from the urine of patients with LCDD recapitulated the biopsy findings (209,210,211,212,213). Human mesangial cells incubated with light chains purified from the urine of patients with LCDD transform from their usual smooth muscle to a myofibroblastic phenotype (214), endowing them with the necessary machinery to engage in active synthesis of extracellular matrix proteins. The initial events were activation of PDGF-β, leading to cellular proliferation through c-fos activation, and later TGF-β activation, resulting in matrix deposition (203,211,212,213). The in vitro model clearly demonstrated the crucial role of TGF-β in the production of extracellular matrix proteins by mesangial cells in LCDD (213). Tenascin is the main extracellular matrix protein in the nodules formed in the in vitro model as well as in the mesangial nodules in nodular glomerulosclerosis in LCDD (197,210,211) by transformed mesangial cells with a myofibroblastic phenotype (214). Tenascin is mainly degraded by matrix metalloproteinase-7 (MMP-7), with minor contributions by MMP-1 and MMP-3 (215). MMP-7 secretion by mesangial cells is impaired in LCDD, making tenascin degradation virtually impossible (216). The specific mechanism responsible for this has not yet been elucidated. The interactions between the light chains responsible for LCDD and mesangial cells occur through a receptor-mediated mechanism (217) (Fig. 22.14).
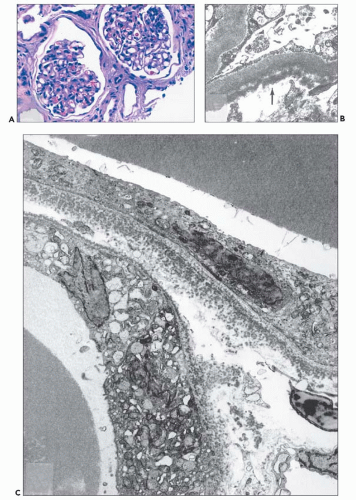 FIGURE 22.13 Light chain deposition disease and light chain cast nephropathy, combined. A: Glomeruli are unremarkable by light microscopy. (H&E, ×500.) B: However, ultrastructurally, deposition of light chain material is noted along the peripheral glomerular capillary walls (arrow) and (C) along the tubular basement membranes, associated with abundant distal tubular casts. B, C: Transmission electron microscopy. (Uranyl acetate and lead citrate.) (B: ×9500; C: ×7500.) |
Although κ light chains are preferentially associated with LCDD, amino acid substitutions introducing hydrophobic residues in the exposed portions of the variable region of the light chains, usually in the CDR1 and CDR3 and less often CDR2 and FR regions, are associated with LCDD, regardless of light chain subtype (218,219). Some light chains with posttranslational glycosylation have also been associated with LCDD (220).
 FIGURE 22.14 Light chain deposition disease. A: Glomerulopathic light chain-mesangial cell interactions with interplay of growth factors—PDGF-β and TGF-β. B: End result in LCDD. Increased mesangial matrix as a result of TGF-β production and decreased activity of metalloproteinases. |
Treatment, Course of the Disease Process, and Prognosis
The outcome in LCDD remains uncertain and depends on how early in its course the disease is detected. Overall, patient survival at 1 and 5 years (89% vs. 70%), respectively, is better than renal survival (67% at 1 year vs. 17% at 5 years) (156). Approximately one third of the LCDD patients without diagnostic features of myeloma develop overt myeloma during the course of the disease, affecting survival adversely. Extrarenal deposits can be asymptomatic or associated with organ damage (182,203).
Treatment is aimed at controlling and reducing the abnormal light chain production by the clone of plasma cells. Chemotherapy with melphalan and prednisone has been employed with success in patients with LCDD and myeloma (221), but the benefit of chemotherapy has been debated when the bone marrow findings are inconclusive, especially if a monoclonal protein cannot be demonstrated in serum or urine (220). Chemotherapy is most effective in cases with a serum creatinine lower than 2 mg/dL. The severity of the underlying plasma cell dyscrasia and the degree of renal failure at presentation, as expected, significantly affect prognosis (89). Bone marrow transplantation has been used, especially in cases detected early, before compromise of other organs has occurred (220,221,222,223). Those patients with overt myeloma do much worse than those with no detectable paraprotein/proliferation of plasma cells and systemic manifestations. However, even in those cases with poor response, progression to end-stage renal disease may take several years.
Early diagnosis and aggressive therapy appear to offer the best hope for these patients in terms of preservation of renal
function and overall survival (223). High-intensity chemotherapy followed by stem cell rescue and bone marrow transplantation has been used in several trials, resulting in increased patients’ survival and improvement in renal function (224), and has become a standard therapy (223).
function and overall survival (223). High-intensity chemotherapy followed by stem cell rescue and bone marrow transplantation has been used in several trials, resulting in increased patients’ survival and improvement in renal function (224), and has become a standard therapy (223).
There are a few reports of resolution or disappearance of nodular glomerulosclerosis after therapy in patients with LCDD, suggesting that early intervention can reverse renal damage in these patients (225,226). New therapies aimed at targeting key steps in the cascade of events involved in glomerular and interstitial damage are to be designed to be used in combination with chemotherapy and other therapeutic interventions that control light chain production (195).
Transplantation in Light Chain Deposition Disease
Renal transplantation has been successful in patients with disease confined to the kidneys (227,228). If the production of the precursor protein is not controlled or eliminated before transplantation, the disease will recur in the transplanted kidney. Because complete control of the plasma cell dyscrasia is virtually impossible, recurrence is inevitable, and recurrence occurs in more than 50% of the patients 8 to 48 months after transplantation and may eventually lead to graft loss (227,228,229,230,231). In a series of patients documenting the outcome of seven LCDD patients that were transplanted, recurrence occurred in five of the allografts (227). Recurrence is not always associated with rapid loss of the transplanted kidney; the median time to reach end-stage renal failure was 33.3 months. One of these transplanted patients was alive with no evidence of recurrence 13 years after transplantation (228). Therefore, renal transplantation can be used to improve quality of life in certain patients but not as a long-term solution in the majority. Unfortunately, even when it is believed that control of the plasma cell clone has been achieved, it can reemerge, followed by light chain deposition. LCDD can recur without clinical evidence of underlying myeloma (231). There is a case report of de novo LCDD arising in a transplanted cadaver kidney 16 years after transplantation (232).
Heavy Chain Deposition Disease and Light and Heavy Chain Deposition Disease
Clinical Presentation and Laboratory Findings
Heavy chain disease (HCD) is a disorder characterized by the production of monoclonal immunoglobulins with truncated heavy chains and no associated light chains. Abnormal heavy chains circulate in the blood of patients with this disorder, and these paraproteins must be identified in the serum and characterized to make a definitive diagnosis. HCD involving three immunoglobulin classes has been described: α-HCD is the most common of the three, followed by γ-HCD and then µ-HCD. Patients with α-HCD present with malabsorption and protein-losing enteropathy; renal disease is not a significant clinical manifestation of this condition and only occurs sporadically. Approximately 150 cases of γ-HCD have been reported, and it is a heterogeneous disorder, which some believe does not justify its diagnosis as a single entity (233). This disease usually manifests with lymphadenopathy, splenomegaly, and constitutional symptoms. Palatal edema and uvula swelling, initially thought to be characteristic of the disease, are only noted in approximately 10% to 15% of all patients with γ-HCD (214). Skeletal involvement occurs in approximately 30% of these patients, and differentiation from myeloma and Waldenström macroglobulinemia (WM) may be difficult on clinical grounds (233). γ-HCD is the most common type of HCD associated with renal manifestations (233). There is an overrepresentation of γ3 and γ4.
A minority of patients with HCD develop tissue deposits, an entity known as HCDD. Bence Jones proteinuria is common. HCD and cast nephropathy have been documented to coexist (151). There is not much experience with µ-HCD and renal disease (171,233,234). HCDD is mainly associated with γ (γ1- and γ3-subtypes) (235,236) and rarely with α-heavy chains (237).
A few hundred patients with HCDD have been described in the literature (155,233). There is no sex predilection for HCDD. Patients with HCDD and renal disease tend to be younger than those with LCDD by a few years (mean age 53 to 55 years), but some patients develop HCDD as early as the third decade of life (156,171). The clinical presentation is similar to that of LCDD patients. Hypertension, nephrotic syndrome, microhematuria, and renal insufficiency are usually present at the time of the renal biopsy (171). Hypocomplementemia is not uncommon in HCDD and correlates with the presence of 1 or 3 subtypes of γ heavy chain (171,236). Signs of complement activation with C3 and C1q deposition in the kidney represent an additional feature of this condition (156,171), mostly when γ1- and γ3-chains are involved (190). Serum hypocomplementemia may also be noted. Hypertension and hematuria appear to be more common in HCDD than in LCDD (171). In some patients with HCDD, a monoclonal component is not detectable in serum and/or urine. Whereas most patients with HCDD have a monoclonal gammopathy, only a few meet the minimal criteria for myeloma (89), at least at presentation.
It is likely that HCDD is underdiagnosed in renal biopsies, and therefore the incidence of this condition is probably underestimated. For example, two recent autopsy series of patients with myeloma (82,88,187) failed to identify any patients with HCDD, suggesting that the overall knowledge about this disease and methods available to diagnose it remain less than optimal. Patients with findings in their renal biopsies that are suggestive of deposition disease with negative stains for κ and λ by immunofluorescence should be worked up for HCDD.
Some cases are combined LCDD and HCDD, termed LHCDD (145), and γ is the most frequent heavy chain component in these patients. Both κ and λ light chains have been found in the LCDD component of these cases (156). The combined variety, known as LHCDD, occurs in older patients—by approximately 10 years—than patients with LCDD or HCDD alone (156). Overall renal function is more significantly impaired at presentation when the two deposition disorders coexist, with an average serum creatinine in the neighborhood of 5 mg/dL. However, proteinuria is usually in the range of 3 g/24 h. The diagnosis of LHCDD requires careful pathologic evaluation of the renal biopsies and a high index of suspicion (156). The diagnosis cannot be reached by light microscopy alone; immunofluorescence with the use of specific antisera is required. Ultrastructural findings may provide confirmatory evidence of light/heavy chain deposition in various renal compartments.
Gross Pathology
There are no studies describing specific gross findings in patients with HCDD.
Light microscopy
In the great majority of the reported cases of HCDD, nodular glomerulosclerosis with features identical to those seen in association with LCDD has been identified (Fig. 22.15) (155,234,235,236,237,238). Crescents were described in four of nine cases, involving 11% to 75% of the glomeruli in one series (155,237). The broad spectrum of glomerular lesions described in LCDD has not been documented in HCDD. A single case of intracapillary proliferative glomerulonephritis has also been reported (239). Crescents are more common in HCDD. Tubular basement membrane deposits with no glomerular or vascular heavy chain deposition have been reported (155). Congo red stain is always negative.
Immunofluorescence
Staining for only one heavy chain class is positive, whereas staining for both light chains is negative. Cases with γ1, γ3, γ4, α, and µ chain deposits have been reported, and γ chain deposits are the most common (156). Thus, cases of HCDD are diagnosed by finding linear staining of renal basement membranes with antisera to one immunoglobulin class (IgG in cases of γ heavy chain, IgA in cases of α heavy chain, or IgM in cases of µ heavy chain), but negativity for both kappa and lambda light chains. The distribution of staining is similar as that for LCDD (Fig. 22.16), but the degree of staining along tubular basement membranes is generally less than in LCDD. The staining for the heavy chain components varies from a predominantly linear to a much less common granular pattern (89,156,178). The heavy chain deposits usually display a uniform, continuous pattern of deposition at the various sites responsible for the linear fluorescence staining pattern noted in the great majority of the cases. Specific antibodies for constant regions of the heavy chain molecule that are deleted in this condition (uniformly lacking CH1 and in some cases also CH2) can be used to demonstrate the absence of these components and confirm the diagnosis (89,125,182,194,195,238). In cases of γ HCDD, staining for the gamma chain subtypes (1,2,3,4) will help to confirm the diagnosis by identifying a single gamma subtype. As previously noted, granular C1q and C3 may be observed in the same distribution as the heavy chain. Extrarenal deposits of heavy chain components have been reported in the pancreas, thyroid, striated muscle, and liver but are less frequent than in LCDD (89,156).
 FIGURE 22.15 Heavy chain deposition disease. Morphologic findings in HCDD are identical to those in LCDD, with nodular glomerulopathy as the typical pattern. (H&E, ×500.) |
 FIGURE 22.16 Heavy chain deposition disease. Intense peripheral capillary wall and mesangial staining, associated with linear tubular basement membrane staining. Direct immunofluorescence staining for µ heavy chain. (Fluorescein, ×350.) |
Electron Microscopy
The ultrastructural findings in HCDD are similar to those in LCDD in most instances. The heavy chain deposits can be subtle or massive. Overall, heavy chain deposits are variable in quantity and distribution in the various renal compartments. In one case, the deposits were described as fibrillary, consisting of 13- to 18-nm-diameter fibrils without periodicity, judged ultrastructurally to be different from those found in fibrillary glomerulopathy. The fibrils exhibited various lengths, with the shorter ones having smooth walls and the longer fibrils exhibiting a “barbed-wire” appearance (240). This case appears to represent an unusual morphologic manifestation of µ-chain HCDD. The patient had no clinical manifestations of Waldenström macroglobulinemia. Another µ-HCDD had massive fibrillary deposits in the mesangium that varied from 16 to 18 nm in diameter and were determined not to be compatible with the material seen in fibrillary glomerulopathy by electron microscopic criteria (234). These two cases suggest that there may be a variant of HCDD with peculiar fibrillary ultrastructural appearance, but this deserves further consideration.
Etiology and Pathogenesis
The deletions in the domains of heavy chain portion of the immunoglobulin molecule (i.e., CH1, CH2, and very rarely, hinge region) result in premature secretion of the heavy chain into the circulation, and these structurally abnormal heavy chains are deposited in target organs, including the kidneys (241). The nascent γ heavy chain protein normally is retained in the endoplasmic reticulum during IgG assembly by binding of its CH1 domain (and to some extent CH2 and hinge region) to chaperone protein, heavy chain-binding
protein. The specific mechanisms involved in the pathogenesis of the renal alterations that occur in this disorder have not been elucidated.
protein. The specific mechanisms involved in the pathogenesis of the renal alterations that occur in this disorder have not been elucidated.
Treatment, Course of the Disease Process, and Prognosis
The therapy employed is similar to that of LCDD, and the results appear to be comparable (198). However, there are no controlled trials addressing the therapy and management of patients with HCDD. Nevertheless, it appears that, based on anecdotal experience, the overall outcome is poor in terms of renal and patient survival. One patient had γ-HCDD for 10 years and subsequently developed γ AL amyloidosis (242).
Transplantation in Heavy Chain Deposition Disease
Differential Diagnosis for Heavy Chain Deposition Disease and Light and Heavy Chain Deposition Disease
Because the light microscopic appearance of LHCDD is so variable, the differential diagnosis includes many diseases, and it must be differentiated from minimal change disease in those cases where the glomeruli appear essentially normal, from mesangial proliferative glomerulonephritis when mesangial proliferation is present, and from membranoproliferative glomerulonephritis, including dense-deposit disease; also, crescentic glomerulonephritis must be distinguished from the proliferative variants of LCDD (108). When considering the most characteristic expression of LCDD and HCDD—nodular glomerulosclerosis—the main differential diagnosis is diabetic nephropathy or “idiopathic” nodular glomerulosclerosis (244). In most instances, demonstration of monoclonal light or heavy chain determinants in the proper histopathologic setting is the essential diagnostic finding. Careful attention to the tubulointerstitial and vascular compartments for light chain deposits is also imperative, as there is a subset of LCDD patients with no glomerular light chain deposition and only tubulointerstitial manifestations of this disease (108,156). A diagnosis of γ-HCDD can be confirmed by using antisera to the IgG subtypes [1-4] and demonstrating staining for a single subtype. It is also helpful to identify deletions of CH1 and, in some cases, CH2 using specific antisera to the constant domains (CH1 to 3) of the γ heavy chain. Rarely, amyloidosis needs to be ruled out, especially in cases with nodular glomerulopathy. In the great majority of these situations, immunofluorescence and electron microscopy suffice, and a solid, unequivocal diagnosis can be obtained. Selective use of immunoelectron microscopy is indicated when the usual diagnostic techniques do not provide enough data to establish an unequivocal diagnosis (157). In the experience of one of the authors (G.A.H.), approximately 5% of the cases require an extended workup beyond routine light, immunofluorescence, and ultrastructural evaluation (157).
Other Entities in the Differential Diagnosis
Glomerulonephritis Associated With Monoclonal IgG Deposits and Monoclonal Gammopathy of Unknown Significance
This entity was first recognized as a specific entity and given its name in 2004 (245). An additional case was reported the same year (246). There are approximately 60 cases of this entity reported in the literature. By light microscopy, these cases exhibit proliferative glomerular changes, most commonly resembling membranoproliferative glomerulonephritis, type I (in about 60% of these cases) (245,246,247,248,249,250) (Fig. 22.17A). The glomerular electron-dense deposits are granular and nonorganized, ultrastructurally resembling immune complex-mediated glomerulonephritis (Fig. 22.17D), but exhibit a unique immunofluorescence profile; they contain monoclonal IgG (predominantly IgG3) and monoclonal light chains (Fig. 22.17BC), either kappa or lambda. The electron-dense deposits are characteristically located subendothelially and also in mesangial areas. In cases with predominantly subepithelial immune complexes, the light microscopic pattern was that of a membranous nephropathy (247,248). Clinically, these patients present with varying degrees of proteinuria (100% of patients reported), which is most often nephrotic range; microhematuria (about 60% of the patients); and renal insufficiency. Another subgroup of these patients exhibit morphologic features that are similar to dense-deposit disease (251).
A monoclonal serum or urine protein is identified in approximately half of the patients; however, only rare patients are associated with myeloma or B-cell lymphoproliferative disorders. The largest series of 37 patients was published in 2009 with only 1 of the patients revealing evidence of myeloma and 1 with AL amyloidosis (250). There are 21 additional patients reported in the literature. Only 3 of these of the 16 patients with available data had a monoclonal protein in the serum and/or urine (245,249,252).
Treatment has consisted of a combination of regimens depending on the particulars of a given case. Renin-angiotensin (RAS) blockade alone was used in about 30% of the patients. Immunomodulatory therapy with or without RAS blockade has been the most commonly used treatment (249). Rituximab has been reported in another series of these patients with satisfactory results (253). It is fair to state at the present time that the best therapy has not been defined due to the small number of patients that have been documented with this condition and the lack of well-defined therapeutic trials.
Short follow-up (<3 years) reported in the largest series demonstrated approximately one third of the patients with complete or partial recovery, another 40% or so with persistent renal dysfunction, and the remainder progressed to end-stage renal disease. One of the patients in this cohort without a monoclonal spike at the time of presentation developed a hematologic malignancy. This entity has been reported to recur in the transplant, where some cases have responded favorably to rituximab (254).
It is important to differentiate these patients from others with monoclonal light chain-related renal disease because this entity does not appear to be a precursor of or significantly associated with an underlying plasma cell dyscrasia, except in rare instances.
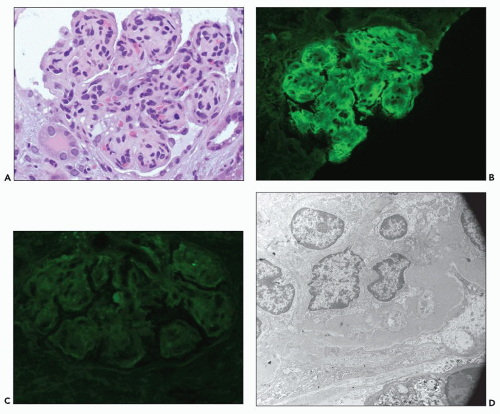 FIGURE 22.17 Proliferative GN with monoclonal deposits. A: Light microscopy with membranoproliferative appearance. (H&E, ×500.) B: Staining along the peripheral capillary walls and in the mesangium. (Immunofluorescence, kappa, ×500.) C: No glomerular staining. (Immunofluorescence, lambda, ×500.) D: Large subendothelial granular electron-dense deposits. (Transmission electron microscopy, uranyl acetate, and lead citrate, ×12,500.) |
Glomerulonephritis With C3 Deposits Associated With Monoclonal Gammopathy of Unknown Significance
The first reported association between isolated C3 deposits and monoclonal gammopathy was by Appel et al. (249). Similar cases have been reported by several groups (251,255,256,257,258). In a group of 81 hepatitis-negative patients with membranoproliferative glomerulonephritis associated with monoclonal light chain deposits, 28 had evidence of monoclonal gammopathy, and 3 had isolated C3 deposits. Nasr et al. (256) reported that 4 adults in a series of 32 patients with C3 deposits and dense-deposit disease had a history of plasma cell dyscrasia. Finally, a series of six patients with MGUS and a glomerulonephritis with isolated C3 staining has been reported more recently (255). These patients range from 40 to 70 years of age, and they presented with hypertension and significant proteinuria with full-blown nephrotic syndrome in three patients. Hematuria was present in the six cases, and all had evidence of renal insufficiency. The renal biopsies demonstrated variable glomerular proliferative features, exudative changes, and some thrombi in capillary spaces. By immunofluorescence, only C3 deposits were apparent along peripheral capillary walls and in the mesangium, indistinguishable from C3 glomerulopathy. No monoclonal light or heavy chain staining was present. Ultrastructurally, there were subepithelial, intramembranous, and mesangial electron-dense deposits. The ultrastructural findings were interpreted as showing overlapping features between type II and III MPGN. In one patient, a repeat renal biopsy demonstrated glomerular monoclonal lambda light chain deposits that colocalized with the C3 in the mesangium and along the peripheral capillary walls. Some of these cases have exhibited features morphologically consistent with dense-deposit disease (258).
Patients were treated with high-dose dexamethasone, either alone or with melphalan or cyclophosphamide. One
patient progressed to myeloma after 11 years of follow-up. Overall, the renal outcome of these patients is poor with progression to end-stage renal disease in most cases (about 80%). Early therapy to control the plasma cell clone may be indicated in at least some of these patients (251).
patient progressed to myeloma after 11 years of follow-up. Overall, the renal outcome of these patients is poor with progression to end-stage renal disease in most cases (about 80%). Early therapy to control the plasma cell clone may be indicated in at least some of these patients (251).
Isolated C3 glomerular deposition, resembling C3 glomerulopathy, likely represents an unusual complication of plasma cell dyscrasias related to the activation of the alternative complement pathway by the monoclonal immunoglobulins. Some of these monoclonal light chains have been shown to interfere with inhibitors of the alternative pathway, such as factor H. The association of C3 glomerulopathy with MGUS, rather than overt, high plasma cell mass myeloma, suggests that prolonged alternative complement pathway activation by the monoclonal immunoglobulins is required for the development of other more advanced glomerular lesions where the monoclonal light chains may appear in renal tissue (259,260,261).
Thus, monoclonal gammopathy should be considered in the differential diagnosis of patients with MPGN. Some of these cases represent examples of LCDD and/or HCDD in stages prior to nodular glomerulosclerosis (109,161,257,258). In these instances, the typical linear immunofluorescence staining along the peripheral capillary walls and in the mesangium, and in outlining tubules and sometimes in vessels, together with punctate, electron-dense material indicative of light chains in the various renal compartments may provide solid diagnostic information. Other cases, however, may represent either proliferative glomerulonephritis with monoclonal IgG deposits or C3 glomerulopathy. Their unique immunofluorescence profiles and ultrastructural features allow differentiation from LCDD and HCDD.
Additional entities to be considered in the differential diagnosis of the above conditions are immunotactoid glomerulopathy and type I (monoclonal) cryoglobulinemic nephropathy. Immunotactoid glomerulopathy has characteristic microtubular structures with variable diameter, but generally in the 30- to 50-nm range identified ultrastructurally. In cryoglobulinemic nephropathy, intracapillary thrombi may be present by light microscopy, and the typically paired and short microtubular structures that characterize cryoglobulins provide a clue at the ultrastructural level. However, there is heterogeneity in the electron microscopic appearance of cryoglobulins, and intraluminal thrombi are not always present, making confirmation of this diagnosis difficult in some cases. Clinical correlation (usually markedly decreased C4 among others) and search for cryoglobulins in the serum may provide valuable confirmatory data. Patients with either proliferative glomerulonephritis associated with monoclonal IgG deposits and those with MGUS and isolated C3 deposits exhibit a significantly lower incidence of dysproteinemia than those with either monoclonal cryoglobulinemic nephropathy or immunotactoid glomerulopathy, making this distinction clinically useful for patient management and prognosis (255).
Plasma Cell Dyscrasias Associated With Crystalline Inclusions in the Glomeruli
HISTORICAL PERSPECTIVE
In 1949, Neuman (262) described groups of small rhomboid crystals distending and deforming the Bowman capsule in the great majority of the glomeruli, along with similar structures in the proximal tubular cells in the kidneys from a patient with myeloma in an autopsy. Similar aggregates of crystals were identified in the lungs and in neoplastic plasma cells in the bone marrow. This initial report was followed by a second autopsy report by Sickel (263) 10 years later of a patient with enlarged kidneys, which on microscopic examination revealed crystalline inclusions in the glomeruli, along with some in the tubular cells. A diagnosis of clinically unsuspected multiple myeloma was made at autopsy. Carstens in 1989 reported a third case with numerous similar inclusions in parietal, visceral, endothelial, and mesangial cells in the great majority of the glomeruli (264). Since then, only rare case reports of this entity have been documented in the literature. While some of these may be included in cases with Fanconi light chain-associated tubulopathy, it is a fact that some patients only present with glomerular clinicopathologic manifestations.
CLINICAL PRESENTATION AND LABORATORY FINDINGS
While the finding of crystalline inclusions in the cytoplasm of tubular cells is a characteristic finding in light chain-associated Fanconi syndrome (discussed previously), and this entity may be sometimes associated with similar inclusions in the glomeruli, macrophages, and plasma cells, a similar entity exists where the crystalline structures are identified predominantly and, in some cases exclusively, in glomerular cells and in the matrix of the Bowman capsule. It is important to recognize this entity, as this may be the presentation of a patient with unsuspected myeloma. In these cases, the clinical presentation is typically proteinuria of variable degree and renal insufficiency, rather than Fanconi syndrome. In at least one reported case, the patient developed renal failure attributed to the glomerular process (264).
This entity is even more rare than Fanconi-associated proximal tubulopathy, with fewer than 10 cases reported in the literature. One of the authors (G.A.H.) has seen three of these cases in biopsy specimens in consultation. One of these cases (265) presented with a clinical picture of thrombotic microangiopathy. The case reported by Carstens was associated with extrarenal amyloidosis diagnosed in a carpal tunnel specimen (264). Another unique feature of this last case is that the glomerular inclusions were detected months before the diagnosis of myeloma was made. In the consultation cases, one had a diagnosis of myeloma, while in the other two, the renal biopsy findings indicated the presence of an underlying plasma cell dyscrasia that was later confirmed.
GROSS PATHOLOGY
LIGHT MICROSCOPY
The main findings are in the glomerular compartment where there is generally mesangial expansion and there may be prominent parietal and epithelial, as well as endothelial cells. The cytoplasm of affected cells is characteristically swollen and sometimes appears vacuolated with rectangularly/rhomboidalshaped empty spaces with angulated borders. The size of the crystalline inclusions is variable, some being quite large. The cellular nuclei may be displaced to the periphery. The one case with clinical manifestations of thrombotic microangiopathy exhibited capillary thrombi composed of crystalline structures,
variably (frequently completely) occluding the lumina but no fibrin (265). The findings are somewhat similar to what is seen in the entity known as “crystal-storing histiocytosis” (152,153); however, the glomerular localization of the inclusions makes this particular entity unique.
variably (frequently completely) occluding the lumina but no fibrin (265). The findings are somewhat similar to what is seen in the entity known as “crystal-storing histiocytosis” (152,153); however, the glomerular localization of the inclusions makes this particular entity unique.
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


