Renal Disease Caused by Hypertension
Jean L. Olson
DEFINITION
Hypertension, or increased blood pressure, is a worldwide problem with a prevalence of greater than 1 billion people (1). The Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7) has published guidelines for the classification of different levels of blood pressure (1) as follows: normal pressure is considered to be less than 120/80 mm Hg; prehypertension is 120 to 139/80 to 89 mm Hg; stage 1 hypertension is 140 to 159/90 to 99 mm Hg; and stage 2 hypertension is greater than 160/100 mm Hg. The percentage of Americans with hypertension has remained at approximately 29% since 2000 according to the 2007-2008 National Health and Nutrition Examination Survey (NHANES) data (2). Control of hypertension has improved from 27.3% in 1988-1994 to 50.1% in 2007-2008 due to increased awareness, treatment of more of the aware patients, and control in more of those who were treated (2).
Hypertension can have a known cause, such as renal artery stenosis, but in most cases, the etiologic factors are unknown, and the condition is deemed “essential” hypertension. The prevalence of the secondary forms of hypertension, in which the cause can be determined, is generally thought to be less than 10% (3). This chapter covers all forms of hypertension but focuses mainly on essential hypertension.
ESSENTIAL HYPERTENSION
Clinical Presentation
Prevalence, Gender, and Age
The global prevalence of hypertension was estimated at 26% in 2005 and is expected to increase to 29% by 2025 (4). The prevalence of hypertension in the United States as determined by an analysis of data from the NHANES was found to be 29% (5). The age-standardized prevalence rate showed an increase from 24% in 2000 (6). Much of this increase can be explained by the increase in overweight/obesity in the population (6). Other factors include improved survival due to effective treatment, increased sodium intake, and a more sedentary lifestyle (6). Men have slightly higher prevalence of hypertension than women. However, in the interval between 2000 and 2005, the prevalence increased in women but not in men (6). Hypertension is a well-known risk factor for chronic kidney disease (CKD) and is currently the second most common diagnosis for patients with prevalent end-stage renal disease (ESRD) (7). However, the percentage of patients with primary hypertensive nephropathy is difficult to determine as such patients are not usually biopsied, and this diagnosis is often made by exclusion.
Although essential hypertension first becomes manifest in middle life, evidence suggests that factors such as low birth weight due to maternal malnutrition may contribute to its development later in life (8,9,10). Small placental size correlates to higher blood pressure even in early childhood (9). Others have related low birth weight and diminished nephron numbers to a higher risk of hypertension in adolescence or adulthood (8,10). The prevalence of hypertension in children has risen in the last decade from a range of 1%-2% in the 1970s to 3%-5% in the early 2000s (11). This increase has occurred
chiefly in essential hypertension and is explained in large part by an increase in the body mass index (BMI) or waist circumference (11,12). Other risk factors include low birth weight, inflammation, elevated uric acid, and high dietary salt intake (12). Childhood hypertension and prehypertension predict increased blood pressure as an adult. Systolic blood pressure rises throughout life with a difference of 20 to 30 mm Hg from early to late adulthood (13). More than half of all individuals aged 60 to 69 are hypertensive, and this proportion increases to 75% of those aged 70 years and older (1). Diastolic blood pressure rises but to a lesser degree and only until the sixth decade (1). In general, cardiovascular risk is related to levels of diastolic pressure in patients under age 60. However, pulse pressure (the difference between systolic and diastolic pressure) is the best predictor of cardiovascular risk in patients older than 60 years as it is a marker of central vascular stiffness (14). Secondary forms of hypertension should be considered in patients less than 30 years of age and in those older than 60 years with sudden onset of hypertension. A careful family history should also be obtained, because many of these forms have a genetic basis.
chiefly in essential hypertension and is explained in large part by an increase in the body mass index (BMI) or waist circumference (11,12). Other risk factors include low birth weight, inflammation, elevated uric acid, and high dietary salt intake (12). Childhood hypertension and prehypertension predict increased blood pressure as an adult. Systolic blood pressure rises throughout life with a difference of 20 to 30 mm Hg from early to late adulthood (13). More than half of all individuals aged 60 to 69 are hypertensive, and this proportion increases to 75% of those aged 70 years and older (1). Diastolic blood pressure rises but to a lesser degree and only until the sixth decade (1). In general, cardiovascular risk is related to levels of diastolic pressure in patients under age 60. However, pulse pressure (the difference between systolic and diastolic pressure) is the best predictor of cardiovascular risk in patients older than 60 years as it is a marker of central vascular stiffness (14). Secondary forms of hypertension should be considered in patients less than 30 years of age and in those older than 60 years with sudden onset of hypertension. A careful family history should also be obtained, because many of these forms have a genetic basis.
Genetic Factors
Essential hypertension is a polygenic quantitative trait with only a small contribution from each gene and with modulation by other genes (epistasis), as well as factors such as gender, race, age, and environment. Epigenetic mechanisms that alter gene expression without changing the DNA sequence of the gene are also at play (15). The genetic contribution to control of blood pressure has been estimated at 31% to 68% (16). The evidence of a genetic effect was first recognized by family and twin studies. Newer approaches include discovery of individual candidate genes, genome-wide linkage studies (GWLS), and genome-wide association studies (GWAS). However, less than 3% of the observed variance can be attributed to the genetic variations found to date (16). Additional information has been provided by the examination of experimental animal models of hypertension (17).
Family and twin studies provided the first evidence of genetic factors in essential hypertension. In one such study, Mo et al. (18) studied 520 offspring with two hypertensive parents, two normotensive parents, or one of each. Those offspring who had two normotensive parents had a mean blood pressure of 121 mm Hg, and 1.3% of them were taking antihypertensive drugs. The intermediate group with one parent of each type showed an average blood pressure of 125 mm Hg, and 2.4% of them used anti-hypertensive drugs. The last group with two hypertensive parents had an average blood pressure of 135 mm Hg, and 11.7% had been prescribed antihypertensive medications. These differences were significant among all three groups. Twin studies are even more powerful because it is likely that any differences are due to environmental influences in the case of monozygotic twins but that of dizygotic twins represent both genetic and environmental differences. Cui et al. (19) found a univariate correlation coefficient of systolic blood pressure of 0.78 between monozygotic twins, 0.50 between dizygotic twins, and 0.12 between spouses.
Investigators at the end of the 20th century expected that they would be able to discover several candidate genes that might be responsible for essential hypertension (20). Candidate gene studies are hypothesis driven as the genes are chosen on the basis of our knowledge of the intersection of pathophysiology of hypertension and potential molecular pathways. Such studies have been successful in finding more than 15 genes responsible for monogenic heritable forms of hypertension (16,21). Many of these will be discussed below in the section on secondary forms of hypertension. The first demonstration of a significant polymorphism in a candidate gene in essential hypertension was in the study by Jeunemaitre et al. (22) of the angiotensinogen (AGT) gene. They found that hypertensive members of sib-pairs in two large populations inherited a variant of AGT with threonine rather than methionine at position 235 (235T) in higher frequency than normotensive members of the pair. Furthermore, these hypertensive patients had higher concentration of AGT in their sera. They concluded that this polymorphism might contribute to the hypertension. The next step was to determine how the polymorphism affects the function of the protein. Hopkins et al. (23) found that homozygous patients with the 235T genotype had a blunted renal vascular response to the infusion of angiotensin II (AII). Studies of candidate genes in essential hypertension have identified over 50 genes in pathways thought to be involved in essential hypertension including the renin-angiotensin system (RAS), renal sodium and water handling, ion channels and cotransporters, kinase regulators, and enzymes and receptors involved in aldosterone synthesis and signaling pathways (Table 20.1) (16,24). However, such studies are limited as they ignore epistasis, geneenvironment interactions, and rare variants. These limitations sometimes lead to conflicting results.
The next development in the determination of the role of specific genes in hypertension was the use of genome-wide linkage scans (GWLS), which allowed the discovery of unsuspected genes and pathways as well as rare variants that had large effects on blood pressure. Most of these GWLS reported associations only, and many were not confirmed (16). A few of these studies were successful with the results confirmed by others. For example, such analysis by Chang et al. (48) found a region on chromosome 1q that contained at least three genes that associated with blood pressure levels. The three genes included ATB1B, RGS5, and SELE. ATB1B encodes the β subunit of the sodium/potassium ATPase, which is involved in sodium
absorption in the kidney, heart contraction, and regulation of vascular smooth muscle tone. RGS5 inactivates the G proteins involved in vasoconstriction that is mediated by angiotensin. It may also invoke vascular remodeling and angiogenesis. SELE affects vasoactive response. Better detection of possible genes can be achieved by the incorporation of age-genetic interactions into the GWLS (49). This method also leads to improved validation. The results of GWLS can be further improved by the use of meta-analysis (50).
absorption in the kidney, heart contraction, and regulation of vascular smooth muscle tone. RGS5 inactivates the G proteins involved in vasoconstriction that is mediated by angiotensin. It may also invoke vascular remodeling and angiogenesis. SELE affects vasoactive response. Better detection of possible genes can be achieved by the incorporation of age-genetic interactions into the GWLS (49). This method also leads to improved validation. The results of GWLS can be further improved by the use of meta-analysis (50).
TABLE 20.1 Candidate genes for essential hypertension | ||
|---|---|---|
|
GWAS were next undertaken in order to find common variants with small effects that are missed in the candidate gene or linkage analyses. Such studies when combined with meta-analysis and huge sample size have defined more than 50 candidate genes that account for less than 3% of blood pressure variability (16). The first two large GWAS meta-analyses were performed in European populations and uncovered 13 new loci for blood pressure traits such as systolic and diastolic blood pressure (51,52). A more recent study by Ehret et al. (53) discovered 16 novel loci also using a European population. Many of these loci were also associated with determination of blood pressure in people of East Asian, South Asian, or African descent (53). Similar methodology studying such traits as pulse pressure and mean arterial pressure uncovered six additional loci (54). Many candidate genes within the loci in all of these studies are reasonable candidates to affect blood pressure regulation or to influence blood pressure directly. Many of the original single nucleotide polymorphisms (SNP) found in these studies have been replicated in later studies (16), indicating the value of such studies.
As the complexity of genetic control of blood pressure regulation has become apparent, the effects of epigenetic mechanisms and epistasis have been revealed. Epigenetics refers to mechanisms whereby environment-gene interactions can occur via DNA methylation or changes to histones that do not alter the DNA sequence yet change DNA expression. These epigenetic modifications occur in response to environmental stressors such as maternal malnutrition and may remain in effect for several generations. Several such epigenetic mechanisms are reviewed by Millis et al. (15). Epistasis, the modulation of the expression of one gene by another, is also being increasingly recognized and complicates the interpretation of candidate gene studies (16,55).
Systems biology including the study of pathways is an emerging area of research that is likely to be critical to our understanding of the genetic control of blood pressure (16,56). The usefulness of this approach is illustrated by an examination of the fibroblast growth factor (FGF) signaling pathway and its relationship to hypertension (57). A GWAS had identified that a particular SNP of FGF1 was associated with essential hypertension in a large Polish kindred and that its product was increased in the kidney in patients with essential hypertension (58) even though this was not a pathway that had originally been thought to be important in blood pressure regulation. These investigators then explored genes in the FGF signaling pathway and found additional allelic variants of genes, namely FGF-binding protein, that also segregated with essential hypertension (57) and were up-regulated in the kidneys of hypertensive patients as compared to controls.
Experimental models of hypertension chiefly in mice and rats have been used to further dissect the genetics of hypertension (55). It is beyond the scope of this chapter to review all of this work, but a few representative examples are given. Cvetkovic and Sigmund reviewed the early use of knockout and transgenic mice (59). Graham et al. (60) applied genome-wide screening to the stroke-prone SHR and then applied congenic/consomic breeding models to confirm the quantitative trait loci and dissect the genes further. The role of experimental models in defining new candidate genes is illustrated by renalase, an amine oxidase that lowers blood pressure (61). Renalase knockout mice are hypertensive as are rats with a small inhibitory mRNA. These experiments prompted a search for SNPs in the renalase gene in human beings. Two such SNPs were found that were associated with essential hypertension (61). Various experimental models of hypertension, genetic, and otherwise have been reviewed by Dornas and Silva (17).
Genetic background may also be used in the future to determine the best therapy for individuals (55). For example, Matayoshi et al. (62) examined polymorphisms in 17 genes and found 2 (one each in the thiazide-sensitive cotransporter gene and adrenergic receptor-beta 3 gene) that were associated with a good therapeutic response to thiazide diuretics that was not seen in those patients without the polymorphisms. Other investigators have begun to study other polymorphisms with an eye to tailoring therapy to the individual patient. This is described further in the section on “Therapy” (see below).
Thus, it is evident that hypertension does not rely on a single gene but is a polygenic complex trait as first predicted by Hamilton et al. (63) in 1954. Although a few single genes may cause hypertension by themselves, most genetic effects act by increasing susceptibility to develop hypertension under certain conditions. Future studies will be aimed at additional gene discovery, dissection of the pathways that lead to regulation of blood pressure and parsing the relationships among the genes and their regulation whether by modulation by other genes, by epigenetic mechanisms, or by interactions with the environment.
Environmental Factors
Several studies have shown clearly that environmental factors are important in the control of blood pressure (1,64,65). Individual factors that have been examined include low birth weight, BMI, physical activity, dietary factors including salt intake, smoking, alcohol consumption, cocaine abuse, use of oral contraceptives (OCs), and psychological stress. The effects of many of these risk factors depend on an interaction between genes and the environment. This interaction is illustrated by the case of intrauterine growth retardation. Decreased nutrition to the fetus can result in the epigenetic changes of altered methylation of DNA or histone acetylation that then may result in changes in gene expression causing variation in the numbers of stem cells committed to different lineages (8,64). The decrease in nephron numbers can lead to hypertension in adult life. Lifestyle risk factors may be beneficial or harmful. In an individual, blood pressure decreases in a linear fashion with weight loss (66). Physical activity or fitness independent of weight loss also reduces blood pressure (66). Zhao et al. (65) reviewed the literature for the effect of 27 dietary factors. They found that 19 were protective and 6 were associated with an increased risk for hypertension. The strongest protective factors were a diet high in fruits and vegetables, the Dietary Approaches to Stop Hypertension (DASH) diet, and to a lesser extent, increased potassium, calcium, magnesium, and polyunsaturated fatty acid intake. The major dietary risk factor was increased sodium intake.
The evidence for this risk is overwhelming. Numerous studies (reviewed by He and MacGregor (67)) have confirmed the risk and have demonstrated that reduction in dietary salt reduces blood pressure. Smoking, alcohol consumption, and cocaine abuse are all known to increase blood pressure acutely (1), but chronic effects are less clear. OCs are also associated with high blood pressure, and this risk is even greater with advancing age, duration of use, and increased body mass (1). Stress may increase blood pressure acutely but does not cause chronic hypertension (68). However, a review of several studies suggestive of this association supports the possibility that a chronic nonadaptive response to stress may result in hypertension (68).
The evidence for this risk is overwhelming. Numerous studies (reviewed by He and MacGregor (67)) have confirmed the risk and have demonstrated that reduction in dietary salt reduces blood pressure. Smoking, alcohol consumption, and cocaine abuse are all known to increase blood pressure acutely (1), but chronic effects are less clear. OCs are also associated with high blood pressure, and this risk is even greater with advancing age, duration of use, and increased body mass (1). Stress may increase blood pressure acutely but does not cause chronic hypertension (68). However, a review of several studies suggestive of this association supports the possibility that a chronic nonadaptive response to stress may result in hypertension (68).
Racial Factors
The prevalence of hypertension in non-Hispanic black men and women is 40.1% compared to 27.4% for non-Hispanic whites (6). The prevalence of hypertension among Mexican Americans is 27.1%. These data are derived from the 1999-2004 NHANES (6). Hypertension begins at an earlier age, is more severe, and is more difficult to control in African Americans (69,70). Blacks also progress to ESRD owing to hypertension 17.7 times more frequently than do whites (71). Parmer et al. (71) examined the effects of high- and low-salt diets on a group of white and black men matched for blood pressure and found an impaired ability to excrete a salt load in the black group. Proposed causes for this altered salt handling in blacks include failure to respond appropriately to AII, reduction in kallikrein-kinin excretion, and hyperinsulinemia (72). As noted earlier, low nephron number has also been associated with the development of hypertension, and blacks as a group do have lower birth weights and likely lower nephron number (10,73). Differences in the pathology of hypertensive-associated renal disease between African Americans and whites have been described (74). Moreover, recent evidence originally suggested that the apparent increase in hypertensive renal disease in African Americans may be secondary to the high frequency of mutations in the nonmuscle myosin heavy chain 9 (MYH9) gene found in this population (69). The strong association between hypertensive African Americans with renal disease and polymorphisms of the MYH9 gene supported the possibility that the renal disease was primary rather than representing hypertensive nephropathy (75). More recently, it has been found that the risk for various forms of CKD in this population is more strongly related to missense mutations in apolipoprotein 1 (APOL1), which is closely linked to MYH9 (76). The functions of APOL1 include among others association with circulating HDL and autophagic pathways; both functions could play a role in hypertensive injury or other forms of kidney disease (76).
Clinical Features
INITIAL PRESENTATION
In the earliest stages, most patients are asymptomatic, and the condition is recognized only at routine examination, when injury to the vessels may already be present. Symptoms that may be recognized include headache, epistaxis, tinnitus, and dizziness, but symptoms are unusual. Excessive weight, smoking, abnormal serum lipid patterns, and diabetes mellitus are common. Repeated blood pressure readings are necessary to establish the diagnosis and to determine the appropriate therapy (77). The prevalence of hypertension is increasing in children. As in adults, these patients do not present with symptoms but are detected on routine evaluation. Hypertension in children is defined as the average of at least three determinations of systolic and/or diastolic blood pressure ≥95% for gender, age, and height (78). Kaelber and Pickett (79) have devised a table to help determine which children should undergo further evaluation following a first reading of elevated blood pressure.
As the duration of hypertension lengthens at any age, evidence for complications may become manifest. The target organs of hypertension include the heart, brain, peripheral arteries, eye, and kidney (80,81). Hypertension accelerates atherosclerosis in the systemic circulation, whereas the lowerpressure pulmonary circulation rarely develops atherosclerotic plaques even in the face of elevated serum lipids (81). The chief manifestations of hypertensive injury in the heart include concentric left ventricular hypertrophy, congestive heart failure, atrial fibrillation, and coronary artery atherosclerosis, leading to increased risk of myocardial infarction (80,81,82). Additional cardiovascular risk is seen in hypertensive patients with the metabolic syndrome (83) defined as three or more of the following conditions: abdominal obesity, triglycerides greater than 150 mg/dL, high-density lipoprotein cholesterol less than 40 mg/dL in men or 50 mg/dL in women, blood pressure greater than 130/85 mm Hg or being treated for hypertension, and a fasting glucose greater than 110 mg/dL. Hypertension is associated with cerebral hemorrhage due to lesions in the penetrating vessels in the midbrain or due to rupture of berry aneurysms. Additional cerebrovascular complications of hypertension include stroke and vascular dementia (80). Aortic aneurysms as well as claudication are well-known peripheral artery manifestations of hypertension. Hypertension may cause retinal arteriolar thickening, hemorrhage, exudates, or papilledema (77). Hypertensive injury to the kidney is manifest chiefly by the appearance of microalbuminuria and/or decline in estimated glomerular filtration rate (eGFR) (84).
ACCELERATED HYPERTENSION (MALIGNANT HYPERTENSION)
The current classification of hypertension no longer uses the term malignant hypertension (1). However, this term remains in the literature. In the past, malignant hypertension was defined as severe elevation of arterial pressure in combination with funduscopic changes including retinal hemorrhages and exudates with or without papilledema (85). It is often accompanied by target organ damage. It affects less than 1% of patients with essential hypertension, and its incidence has not declined since its description (86). The clinical symptoms may include visual disturbances, headache, headache with visual disturbance, heart failure, stroke or transient ischemic attack, or dyspnea (87). Hematuria is present in as many as 21% of patients, and some patients have gross hematuria. Significant proteinuria is present in 63.4% of patients and was associated with higher serum creatinine levels (87).
Malignant hypertension may occur without any history of hypertension, or it may be preceded by a period of essential hypertension. It may also complicate secondary forms of hypertension such as renal parenchymal disease, renal artery stenosis, various endocrinologic causes of hypertension, or cocaine abuse (86,88). Lane et al. (86) studied 446 patients (64.5% were men) with malignant hypertension who had been admitted to or were referred to City Hospital, Birmingham, England, between 1964 and 2006. Of these patients, 54.3%
had a history of essential hypertension. The remaining patients had secondary hypertension caused by renal artery stenosis, renal parenchymal diseases, or other such conditions. They found an incidence of 1 to 2 cases of malignant hypertension per 100,000 per year and found no apparent decline over the 40 years of the study. An excess of black and Asian patients has been noted relative to the number expected for the population studied in several cohorts (86,89). Survival has increased with the introduction of more effective antihypertensive drugs. In particular, patient survival has increased from 33% to 90% over the past 40 years (86) with 10-year renal survival of 84% (90). Factors involved in determining survival include creatinine at presentation and successful control of blood pressure and proteinuria during follow-up (86,90).
had a history of essential hypertension. The remaining patients had secondary hypertension caused by renal artery stenosis, renal parenchymal diseases, or other such conditions. They found an incidence of 1 to 2 cases of malignant hypertension per 100,000 per year and found no apparent decline over the 40 years of the study. An excess of black and Asian patients has been noted relative to the number expected for the population studied in several cohorts (86,89). Survival has increased with the introduction of more effective antihypertensive drugs. In particular, patient survival has increased from 33% to 90% over the past 40 years (86) with 10-year renal survival of 84% (90). Factors involved in determining survival include creatinine at presentation and successful control of blood pressure and proteinuria during follow-up (86,90).
The cause of the switch from benign to malignant hypertension is not yet fully understood. Many authors have suggested that the renin-angiotensin axis may be paradoxically stimulated by an ischemic renovascular bed stemming from microvascular damage due to increased blood pressure (91,92). This theory has been supported by a transgenic model of malignant hypertension that has been developed by the insertion of the mouse Ren-2 renin gene into Sprague-Dawley rats (93). The affected rats had increased blood pressure and typical renal changes of malignant hypertension and died at 50 to 90 days of age. Heterozygotes showed target organ damage in the heart and kidney but no other characteristics of malignant hypertension. This model has been further refined by inserting the Ren-2 renin gene fused to the cytochrome P450 1a1 (Cyp1a1) promoter (94) that allows controlled inducible angiotensin II-dependent hypertension. Study of this model shows the expected pathologic findings of malignant hypertension and allows therapeutic manipulation. For example, administration of aliskiren, which directly inhibits renin, normalizes blood pressure in these rats (95). Patients with malignant hypertension have elevated plasma renin activity and aldosterone. These levels correlate to markers for intravascular hemolysis and renal function in patients with malignant hypertension (92).
Laboratory Findings Including Proteinuria
Hemolytic anemia is frequently seen in malignant hypertension, with schistocytes on the peripheral blood smear. Thrombocytopenia and negative results on Coombs test are also part of this hematologic picture, which is also known as microangiopathic hemolytic anemia or thrombotic microangiopathy (TMA). Other conditions associated with this condition include hemolytic-uremic syndrome, thrombotic thrombocytopenic purpura, scleroderma renal crisis, and various drug-related injuries. These conditions are described in Chapters 18 and 25. Hyperuricemia has been associated with an increased risk in the future for hypertension particularly in the young and in women independent of other risk factors (96).
Most of the abnormal laboratory values in patients with hypertension reflect end-organ damage. When microalbuminuria is defined as 20 to 200 µg/min (30 to 300 mg/24 hours), the prevalence of microalbuminuria ranges between 5% and 60% in hypertensive patients (97,98). The microalbuminuria correlates to the level of arterial pressure. Furthermore, microalbuminuria is predictive of renal damage, cardiovascular disease, and death in hypertensive patients (80,97,99,100). Wang et al. (101) examined the urinary albumin-to-creatinine ratio in 1499 nonhypertensive individuals. During a mean follow-up period of 2.9 years, 15% of these persons developed hypertension and another 33% progressed to a higher level of hypertension. The authors found that urinary albumin excretion (UAE) predicted these changes in blood pressure even at levels lower than the conventional threshold for microalbuminuria. Microalbuminuria arises in part due to endothelial dysfunction as is discussed below. Microalbuminuria is also secondary to changes in glomerular hemodynamics due to loss of autoregulation. Ten percent of patients at the onset of their hypertension have hyperfiltration (99).
Biomarkers other than microalbuminuria have also been studied in hypertension. The biomarkers studied fall into the categories of markers of endothelial dysfunction, oxidative stress, and inflammation. Endothelial damage is known to occur and may precede the elevated UAE (97). Microalbuminuric essential hypertensive patients had higher serum levels of circulating endothelin-1 (ET-1) and basic FGF (102). Furthermore, there was a correlation between these factors and the level of microalbuminuria. Pedrinelli et al. (103) examined levels of von Willebrand factor (vWF) in 10 hypertensive patients with microalbuminuria and compared them with 10 hypertensive patients without microalbuminuria and 10 controls without microalbuminuria. These investigators found that vWF was increased in the serum of patients with microalbuminuria. Furthermore, serum levels of vWF correlated with UAE, mean blood pressure, and age. Mean blood pressure and vWF both contributed to the rise in UAE when multiple regression analysis with UAE as the dependent variable was performed. Patients with microalbuminuria had higher serum creatinine levels and lower creatinine clearance than did hypertensive patients without microalbuminuria. An inverse correlation was found between vWF and creatinine clearance. The authors concluded that blood pressure may contribute to endothelial damage and thereby may promote microalbuminuria. Other investigators found that serum levels of both intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 were increased in patients with essential hypertension as compared to controls (104). Furthermore, the levels were highest in such patients with microalbuminuria. Additional markers of endothelial dysfunction include inhibitors of nitric oxide (NO) such as plasma asymmetric dimethylarginine and homocysteine. Markers of inflammation such as C-reactive protein (CRP) and plasminogen activator inhibitor-1 may also be elevated. Any of these indicators may be found in patients with hypertension and even in some patients with white coat hypertension (105). Of further interest is that the markers of inflammation including CRP and tumor necrosis factor-α may be present in patients with prehypertension (106,107).
Biomarkers can also be used for risk stratification for end-organ damage, especially the heart, due to hypertension. A recent systematic review and meta-analysis showed that elevated levels of matrix metalloproteinases (MMP)-2 and 9 as well as tissue inhibitor of MMP (TIMP) are present in hypertensive patients (108). MMP-9 and TIMP are elevated in hypertensive patients prior to the onset of heart failure and may be good markers of potential risk for end-organ damage. TIMP has higher levels in patients with left ventricular hypertrophy, indicating the presence of myocardial remodeling. Serum MMP-2 is elevated to a greater degree in hypertensive patients with diastolic heart failure (108). Other useful biomarkers of ventricular remodeling include atrial natriuretic peptide and brain natriuretic peptide (109).
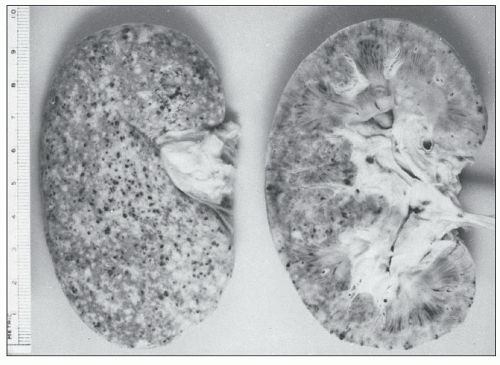 FIGURE 20.1 Kidney from a patient who had the malignant phase of essential hypertension. Kidney weights: left, 150 g; right 130 g. Note numerous petechial hemorrhages. |
Pathologic Findings
Gross Pathology
The kidneys in benign nephrosclerosis are typically reduced in size, with a total weight ranging between 120 and 250 g (normal = 300 g total weight). The two kidneys are usually affected equally. The capsular surface is most commonly finely granular, reflecting disease in small arteries and arterioles. The granules are formed by zones of relatively preserved or even hypertrophied renal parenchyma alternating with neighboring nephrons with diseased arterioles, which are scarred and depressed. When larger renal arteries show intimal thickening and damage larger clusters of nephrons, however, coarser granularity may be superimposed against a background of fine granularity with scattered V-shaped pits. Such larger scars will take the form of pits, which, on cut surface, extend only through the cortex and not the medulla, indicating their relationship to the interlobular or arcuate vessels. These features differentiate them from pyelonephritic scars, which are larger and U shaped, and extend through a fibrotic medulla to end in a dilated, distorted, and inflamed calyx. The capsule may strip with difficulty over such pitted scars. The kidney may be difficult to cut because of the interstitial fibrosis. The cut surface also reveals cortical thinning. Glomeruli may be difficult to identify. Atherosclerosis may be evident in the main renal artery or in any of its branches. Cortical cysts or small adenomas may also be present. If the patient has a short history of hypertension or only mild elevations, there may not be any gross alterations.
The gross appearance of the kidney from an untreated patient with accelerated hypertension differs from that of a patient who has been treated or who has had a period of less severe hypertension preceding the onset of malignant hypertension. In the untreated patient, the combined kidney weights can range from 130 to 410 g (91). The capsular surface may be smooth in those patients without preceding hypertension but may show a granular surface for those with long-standing hypertension. Petechial hemorrhages, frequently representing congested glomeruli sometimes with hemorrhage into local tissue, are prominent and may also be seen on the cut surface. Small infarcts may also be noted (Fig. 20.1). In patients who have been treated, the petechial hemorrhages and infarcts are less prominent or not visible at all.
Light Microscopy
GLOMERULI
The glomeruli may be normal or may simply show age-related changes (110). A few globally sclerosed glomeruli are seen in all adults despite the absence of increased blood pressure. Sclerosed glomeruli may disappear and sometimes one may find such “disappearing glomeruli” that appear to merge into the surrounding renal parenchyma (Fig. 20.2). In hypertensive nephropathy two additional types of glomerular changes are seen: ischemic glomeruli and solidified ones (111). The earliest change is that of collapse of the capillary loops with apparent thickening of their walls on hematoxylin and eosin
(H&E)-stained sections. Periodic acid-Schiff (PAS) reaction or silver stain, however, shows that the apparent thickening is chiefly due to wrinkling of the capillary basement membrane (Fig. 20.3). With time, the entire glomerular tuft shrinks and retracts to the vascular pole. This is accompanied by the filling in of the Bowman space by a faintly eosinophilic material. H&E-stained sections of such glomeruli reveal only pink, nondescript balls (Fig. 20.4). However, PAS staining demonstrates the shrunken tuft as well as mild thickening of the Bowman capsule (Fig. 20.5). Furthermore, the material filling the Bowman space is PAS negative, a characteristic staining pattern for collagen. This change may begin in the hilar portion of the tuft, but it soon fills the entire space emptied by the shrinking glomerulus (112) until it becomes obsolescent.
(H&E)-stained sections. Periodic acid-Schiff (PAS) reaction or silver stain, however, shows that the apparent thickening is chiefly due to wrinkling of the capillary basement membrane (Fig. 20.3). With time, the entire glomerular tuft shrinks and retracts to the vascular pole. This is accompanied by the filling in of the Bowman space by a faintly eosinophilic material. H&E-stained sections of such glomeruli reveal only pink, nondescript balls (Fig. 20.4). However, PAS staining demonstrates the shrunken tuft as well as mild thickening of the Bowman capsule (Fig. 20.5). Furthermore, the material filling the Bowman space is PAS negative, a characteristic staining pattern for collagen. This change may begin in the hilar portion of the tuft, but it soon fills the entire space emptied by the shrinking glomerulus (112) until it becomes obsolescent.
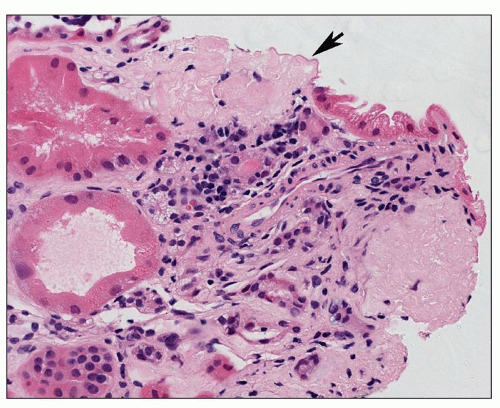 FIGURE 20.2 Disappearing glomerulus (arrow) merging into surrounding tissue. (H&E, ×260.) |
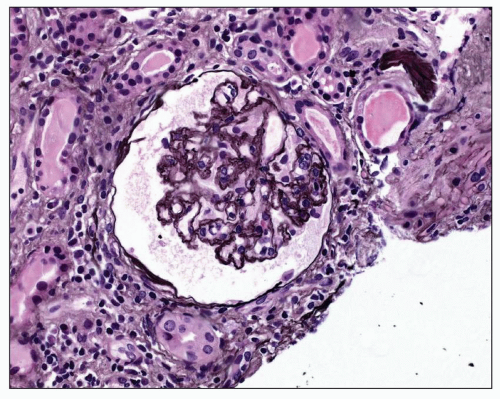 FIGURE 20.3 Glomerulus with wrinkling of the GBM accompanied by reduction of capillary lumen diameter. (PAS-Jones, ×390.) |
The solidified glomerulus is characterized by an increase in the mesangial matrix, which results in either segmental or global solidification (sclerosis) of the glomerular tuft (Fig. 20.6), extending to the Bowman capsule without collagenization of the Bowman space. Such glomeruli frequently contain hyalinosis lesions and represent “decompensated benign nephrosclerosis” (DBN) (113,114). This term is used by Bohle et al. to describe those cases of benign nephrosclerosis in which many glomeruli show prominent hyalinosis accompanied by mesangial widening. In a study of hypertensive patients, these authors found that 775 kidneys showed the obsolescent glomeruli described above accompanied by hyaline arteriolosclerosis. An additional 251 patients had the solidified glomeruli of DBN (114). These latter patients developed chronic renal failure within a few years and had higher levels of blood pressure, proteinuria, and higher
serum creatinine than patients with solely ischemic lesions. Solidified glomeruli are readily distinguished from obsolescent glomeruli by use of the PAS staining technique. Marcantoni et al. (74) recognized these solidified glomeruli of DBN in a higher proportion of African American hypertensive patients than in Caucasians. They also reported a higher frequency of segmental sclerosis in the patients with DBN. Thus, this alteration may represent a secondary form of focal segmental glomerulosclerosis (FSGS). Kincaid-Smith (115) has suggested that the increase in segmental glomerular lesions may be related to the increase in the association between the metabolic syndrome (chiefly obesity and insulin resistance) and hypertension. She posits that these factors are producing the segmental glomerular sclerosis rather than the hypertension. Kidneys with hypertensive nephrosclerosis also may contain a population of hypertrophied glomeruli (116). Hill et al. (116,117) have described pathologic differences in the afferent arterioles supplying these different types of glomeruli. This is addressed below in the discussion of the pathogenesis of these changes.
serum creatinine than patients with solely ischemic lesions. Solidified glomeruli are readily distinguished from obsolescent glomeruli by use of the PAS staining technique. Marcantoni et al. (74) recognized these solidified glomeruli of DBN in a higher proportion of African American hypertensive patients than in Caucasians. They also reported a higher frequency of segmental sclerosis in the patients with DBN. Thus, this alteration may represent a secondary form of focal segmental glomerulosclerosis (FSGS). Kincaid-Smith (115) has suggested that the increase in segmental glomerular lesions may be related to the increase in the association between the metabolic syndrome (chiefly obesity and insulin resistance) and hypertension. She posits that these factors are producing the segmental glomerular sclerosis rather than the hypertension. Kidneys with hypertensive nephrosclerosis also may contain a population of hypertrophied glomeruli (116). Hill et al. (116,117) have described pathologic differences in the afferent arterioles supplying these different types of glomeruli. This is addressed below in the discussion of the pathogenesis of these changes.
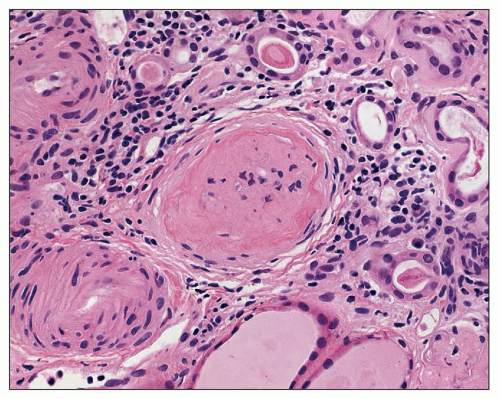 FIGURE 20.4 Sclerotic glomerulus with obliterated capillary loops. (H&E, ×260.) |
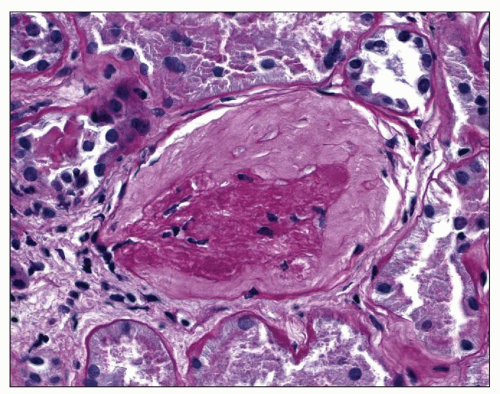 FIGURE 20.5 The glomerular tuft is shrunken with wrinkling of the capillary walls. The capsular space contains pale-staining material. The PAS technique clearly distinguishes between the collapsed tuft and the collagenous material occupying the capsular space. (PAS, ×340.) |
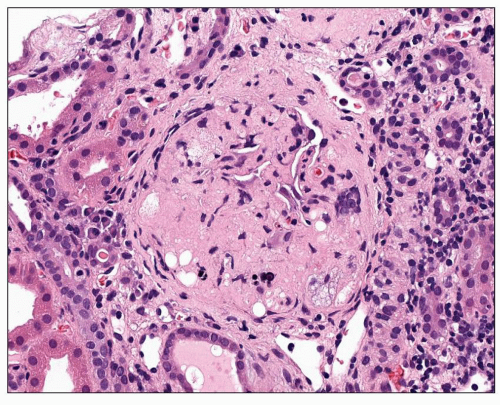 FIGURE 20.6 Solidified glomerulus extending to the Bowman capsule without collagenization of the Bowman space. Foam cells may be seen within the glomerulus. (H&E, ×260.) |
Several other glomerular alterations have been noted in the kidneys from hypertensive patients. First, the podocyte number is decreased in hypertensive nephrosclerosis accompanied by increased urinary podocin, nephrin, and synaptopodin as compared to healthy controls (118). Reduced podocyte numbers have been described in a number of renal diseases with glomerulosclerosis. Hoy et al. (119) have demonstrated lower glomerular number in hypertensive American whites and Australian Aborigines. However, there was no difference in nephron number in hypertensive blacks from the United States as compared to nonhypertensive blacks. However, the mean glomerular volume was greater in hypertensive blacks as compared to nonhypertensive controls (119). Finally, the juxtaglomerular apparatus (JGA) is slightly enlarged with respect to numbers of cells in patients with benign hypertension compared with normotensive controls (120).
The glomerular changes in accelerated (malignant) hypertension may be acute or chronic (121,122,123). They are identical to those seen in secondary forms of malignant hypertension in association with a preexisting renal parenchymal disease; the alterations described here are superimposed on the changes of the respective condition. The acute changes are focal; most glomeruli appear unchanged. The most obvious change is that of fibrinoid necrosis, which is usually segmental (Fig. 20.7). This lesion is eosinophilic, but it is most easily seen with a silver stain, which demonstrates interruption of the glomerular basement membrane (GBM). Furthermore, the intense eosinophilia of the area of necrosis stands in sharp contrast to the black staining of the GBM and mesangial matrix (Fig. 20.8). The mesangial matrix may also show loosening, so-called mesangiolysis. The fibrinoid necrosis may extend from the afferent arteriole, or it may be peripheral and associated with a crescent. Fibrin, which accounts for the eosinophilia of the lesion, may also be seen in other capillary lumina within the glomerular tuft or may accumulate under endothelial cells. Endothelial cells of arterioles and capillaries may become swollen and obscure the lumen. Fibrin platelet thrombi with red blood cell fragments may also be seen (Fig. 20.9). In other glomeruli, the predominant alteration is one of intense congestion with dilation of the capillary lumina (Fig. 20.10). The final acute alteration is that of thickening of capillary walls in open capillary lumina. Silver stain demonstrates double contours indicative of subendothelial widening and new basement membrane formation (Fig. 20.11) (see also Chapter 18)
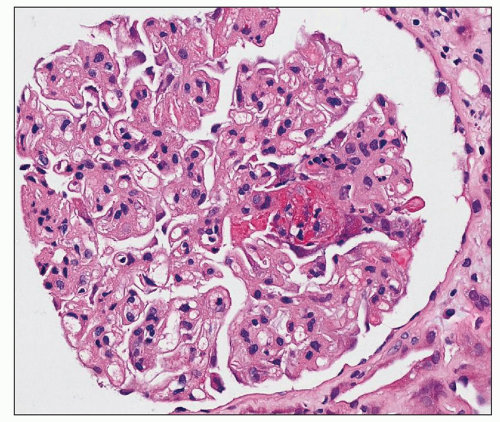 FIGURE 20.7 Glomerulus with segmental necrotizing lesion (fibrinoid necrosis) characterized by eosinophilia, karyorrhexis, and apparent disruption of glomerular architecture. (H&E, ×260.) |
The chronic changes are of two different types. The first is similar to that seen in benign hypertension, in that there is collapse of the tuft with wrinkling of the GBM accompanied by collagenization of the Bowman space. This change may occur with or without a preceding period of benign hypertension, although it is much more prominent in patients with a history of hypertension. In the second type, glomeruli are also collapsed, but seem almost acellular and, on silver stain, show the subendothelial widening described earlier in the preceding paragraph. Thus, the collapsed glomeruli have evolved from those that showed subendothelial widening.
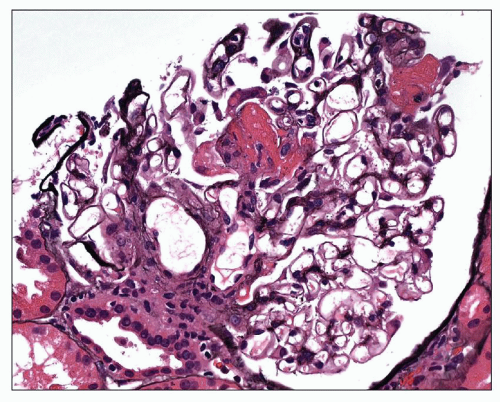 FIGURE 20.8 Fibrinoid necrosis involving several lobules in the center and upper right of the tuft, with segmental disruption of the basement membrane at the central portion. (PAS-Jones, ×260.) |
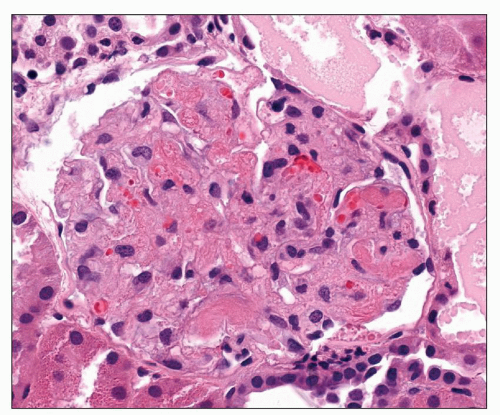 FIGURE 20.9 Glomerulus with dilated capillaries filled with red cells, red cell fragments, and amorphous material probably representing platelets and fibrin. An identical picture is seen in other conditions with TMA such as hemolytic-uremic syndrome and scleroderma. (H&E, ×520.) |
TUBULES
The tubules in either benign or malignant hypertension may be atrophic and sometimes contain hyaline casts. The epithelial cells are flattened and surrounded by a thickened tubular basement membrane, which may be wrinkled. Such atrophic areas often alternate with zones of tubules with dilated lumina and tall hypertrophied epithelial cells (Fig. 20.12). Such zones form the granularity seen on the capsular surface of gross specimens. The size of the scars, or fields of atrophic tubules, depends on the diameter of the narrowed vessel supplying the area, thus determining the number of nephrons affected. Although such atrophic tubules are frequently near an obsolescent glomerulus, this is not always the case, a finding that may reflect the greater susceptibility of tubules to ischemia. Actual loss of tubules may also occur suggested by glomerular crowding. If the patient has proteinuria, protein reabsorption droplets may be present in the tubular epithelial cells. In patients with sudden onset of malignant hypertension, patchy necrosis of tubules may be evident, and there may be cortical infarcts. Such areas are more common in scleroderma-associated TMA than in that associated with malignant hypertension (see also Chapter 18.)
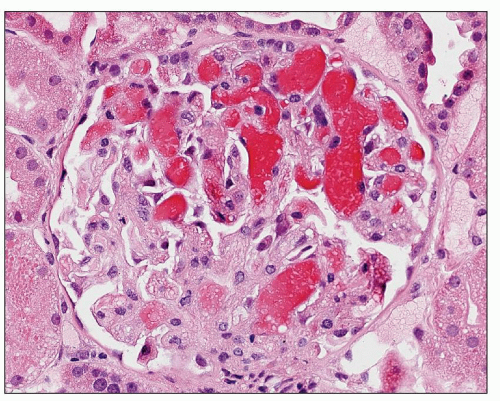 FIGURE 20.10 Glomerulus with congested capillary loops. (H&E, ×260.) |
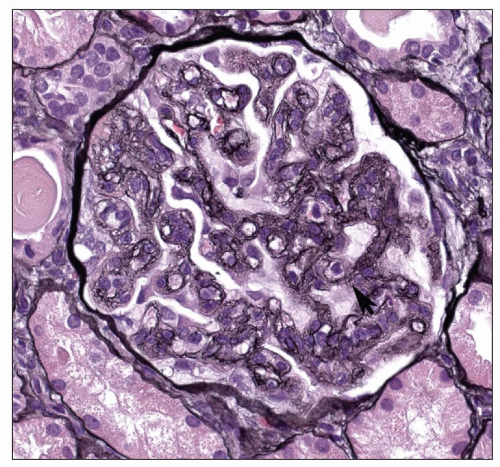 FIGURE 20.11 Glomerulus with double contours (arrow) representing duplication of the GBM and subendothelial widening. (PAS-Jones, ×370.) |
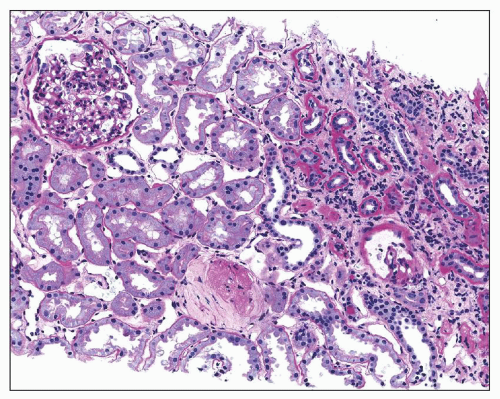 FIGURE 20.12 Renal biopsy showing the zone of atrophic tubules and interstitial fibrosis to right with normal tubules and the interstitium to left. Some of the normal tubules show mild dilation. (PAS, ×125.) |
INTERSTITIUM
The interstitium is widened in areas with atrophic tubules. Increased collagen is noted. Chronic inflammatory cells, usually small lymphocytes, may be widely dispersed in the areas of scarring; however, larger aggregates of lymphocytes may be present in the scars in subcapsular zones. Mast cells are increased in the interstitium early in hypertensive nephropathy (124). Morphometric studies by Grund et al. (125) have shown a correlation between the amount of interstitium and the serum creatinine level. These authors hypothesized that the renal intertubular capillaries had diminished cross-sectional area resulting from interstitial fibrosis and that this change may have contributed to the decrease in the GFR. They did not determine whether the fibrosis preceded the loss of capillaries or vice versa. The chronic hypoxia hypothesis as a model for the progression of renal disease was first suggested by Fine et al. (126). One of the mechanisms for this is damage to the tubules due to reduced blood flow through the peritubular capillaries (111). The mechanisms are further discussed below. The changes in accelerated hypertension are similar to those seen in benign nephrosclerosis.
BLOOD VESSELS
The changes vary with the size of the vessel involved and also differ among individual patients. Arcuate and larger arteries show the alterations typical of atherosclerosis, as manifest by fibrous intimal thickening that results in reduction of the vessel lumen. The internal elastic lamina may show splitting, which is best seen with either a silver or elastic stain. This change is different from the abrupt breaks seen in vasculitis. Lipidcontaining macrophages are not usually evident except in the intima of the main renal artery or its branches. In cases in which malignant hypertension is primary, and not secondary to a period of lower levels of hypertension, the larger arteries may not show any alteration. Otherwise, the arteries are similar in appearance to those seen with any level of hypertension.
Interlobular arteries show fibroelastic intimal thickening with reduplication of the internal elastic lamina (Fig. 20.13). The degree of intimal thickening in arteries is closely linked to this change in arterioles (127). The media may be thickened in small arteries and/or arterioles (Fig. 20.14), reflecting hypertrophy, hyperplasia, or remodeling. In any case, lumen size is decreased, and this decrease may be the result of remodeling, alterations in stiffness or compliance, and functional abnormalities. Remodeling and functional changes are discussed below. Stiffness may be dependent on alterations in the amount and type of collagen, the collagen-elastin ratio, and the presence of proteoglycans (128). The latter has been described in humans. An abnormal interaction among these elements may also lead to increased stiffness (128). In patients with accelerated hypertension, the smaller of the interlobular arteries may also show fibrinoid necrosis, which is a segmental change causing localized destruction of a vessel (Fig. 20.15). However, thickening of the intima with mucoid matrix and widely spaced, concentrically arranged cells are more typical changes (Figs. 20.16). The matrix has sparse collagen in a basophilic edematous-appearing
matrix. This thickened intima may also show lipid-containing macrophages as well as reduplication of the elastic lamina (Fig. 20.17). Elastic reduplication is particularly prominent in those patients who had a prolonged period of benign hypertension before the onset of malignant hypertension. In these cases, the intimal thickening is luminal to the elastic reduplication. The intimal thickening extends along most of the length of the vessels and causes marked narrowing of the lumen that results in the devastating ischemic alterations that can cause renal failure. The most common terms used to describe this lesion are onionskin thickening, endarteritis fibrosa, and musculomucoid intimal thickening. These alterations in the kidney in malignant hypertension, including the findings of fibrinoid necrosis of the afferent arteriole, glomerulitis, and onionskin thickening of small arteries, were first described by Volhard and Fahr (129).
matrix. This thickened intima may also show lipid-containing macrophages as well as reduplication of the elastic lamina (Fig. 20.17). Elastic reduplication is particularly prominent in those patients who had a prolonged period of benign hypertension before the onset of malignant hypertension. In these cases, the intimal thickening is luminal to the elastic reduplication. The intimal thickening extends along most of the length of the vessels and causes marked narrowing of the lumen that results in the devastating ischemic alterations that can cause renal failure. The most common terms used to describe this lesion are onionskin thickening, endarteritis fibrosa, and musculomucoid intimal thickening. These alterations in the kidney in malignant hypertension, including the findings of fibrinoid necrosis of the afferent arteriole, glomerulitis, and onionskin thickening of small arteries, were first described by Volhard and Fahr (129).
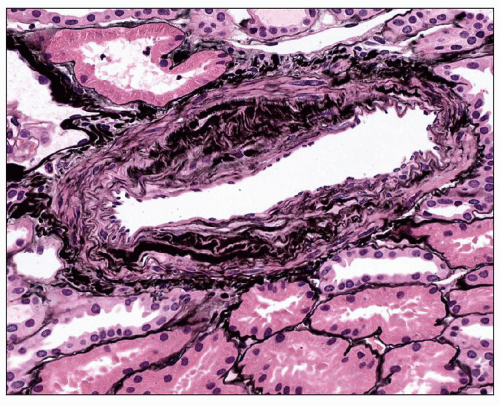 FIGURE 20.13 Interlobular artery with reduplication of the internal elastic lamina. (PAS-Jones, ×260.) |
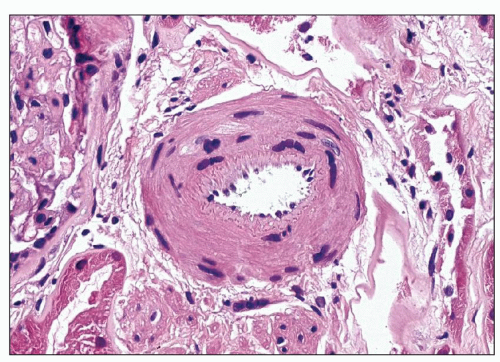 FIGURE 20.14 Small artery showing medial thickening with multiple layers of smooth muscle cells and wrinkling of the internal elastic lamina. (H&E, ×325.) |
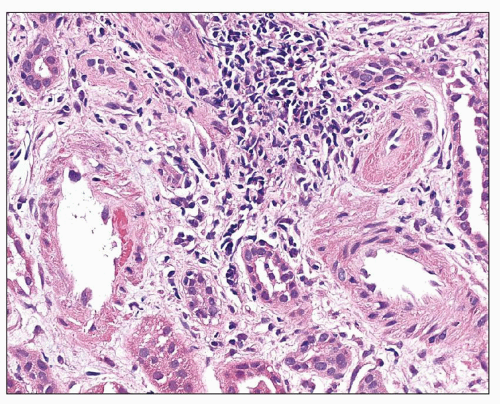 FIGURE 20.15 Small artery to left showing segmental fibrinoid necrosis characterized by intense eosinophilia and granular texture. Arteriole at upper right shows fibrinoid necrosis with near occlusion of the lumen. (H&E, ×260.) |
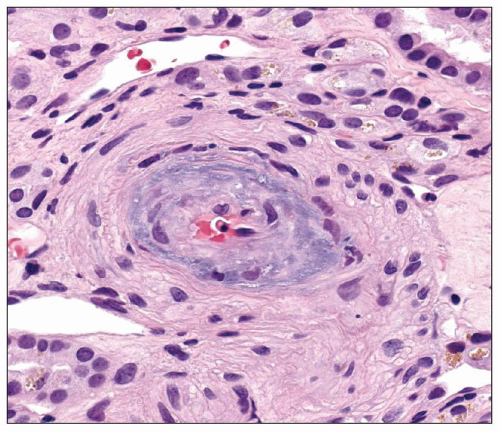 FIGURE 20.16 Small artery with mucinous basophilic intimal thickening. (H&E, ×470.) |
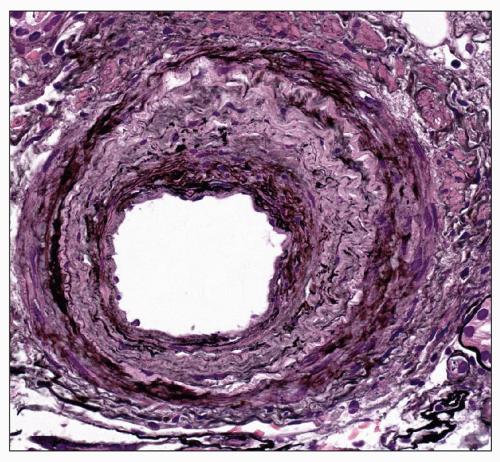 FIGURE 20.17 Interlobular artery with marked reduplication of the internal elastic lamina. (Periodic acid-methenamine silver, ×170.) |
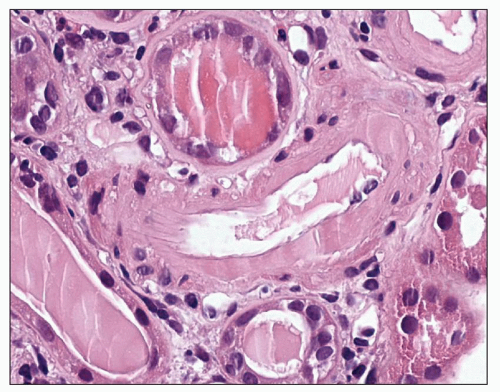 FIGURE 20.18 Arteriole with circumferential intimal hyalin deposition. Note glassy appearance. (H&E, ×520.) |
Arterioles are commonly affected by hyaline arteriolosclerosis, a change that is also present in the aging kidney. Hill (111) prefers the term arteriolar hyalinosis as the hyaline deposits occur in areas of loss of smooth muscle or arteriomalacia. Burchfiel et al. (130) found that the risk of arteriolar hyalinization increased with increases in diastolic blood pressure. The severity of arteriolar hyalinization also correlates with the degree of larger artery intimal thickening (131). This lesion is characterized by the presence of homogeneous eosinophilic material, sometimes with lipid in the subendothelium of the arterioles (Fig. 20.18). It may extend into the media and is more easily seen using the PAS stain (Fig. 20.19). Use of the
term hyalin suggests that it appears glassy, to distinguish it from the more granular texture of fibrinoid necrosis, a lesion typical of the malignant phase of hypertension discussed below. In a morphometric study, Hill et al. (116) noted that arteriolar hyalinosis increased in frequency in hypertensive kidneys as compared to age-matched controls without hypertension. Furthermore, these vessels had larger luminal diameters and an increase in vascular smooth muscle and the extracellular matrix, resulting in a larger outer circumference. The afferent arterioles with hyalinosis were associated with either hypertrophied glomeruli or glomeruli with FSGS. These changes reflect the loss of autoregulation that may occur with hypertension resulting in transmission of elevated pressure through the dilated vessels and on to the glomerulus.
term hyalin suggests that it appears glassy, to distinguish it from the more granular texture of fibrinoid necrosis, a lesion typical of the malignant phase of hypertension discussed below. In a morphometric study, Hill et al. (116) noted that arteriolar hyalinosis increased in frequency in hypertensive kidneys as compared to age-matched controls without hypertension. Furthermore, these vessels had larger luminal diameters and an increase in vascular smooth muscle and the extracellular matrix, resulting in a larger outer circumference. The afferent arterioles with hyalinosis were associated with either hypertrophied glomeruli or glomeruli with FSGS. These changes reflect the loss of autoregulation that may occur with hypertension resulting in transmission of elevated pressure through the dilated vessels and on to the glomerulus.
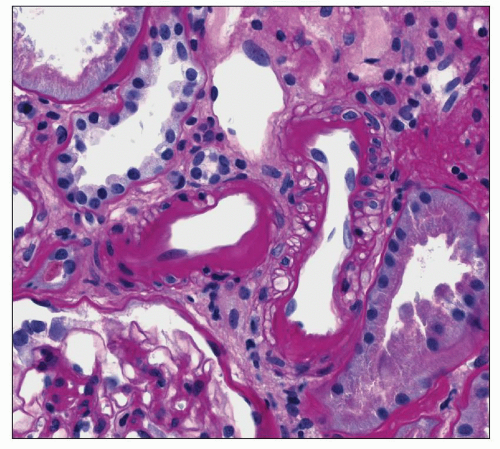 FIGURE 20.19 Small artery with intimal hyalin and arteriole to left with intimal deposition of hyalin extending toward the media and compressing smooth muscle cells. (PAS, ×400.) |
The characteristic lesion of severe (malignant) hypertension is fibrinoid necrosis. Eosinophilic material with a granular texture supplants the medial smooth muscle cells with loss of cell nuclei (see Fig. 20.15). This granular material must be differentiated from the glassy appearance of arteriolar hyalinosis, the more typical lesion of benign hypertension (see Fig. 20.18). Of course, arteriolar hyalinosis may also be present in patients with malignant hypertension, but the identification of true fibrinoid necrosis suggests the diagnosis of the accelerated phase. Fibrinoid necrosis also impinges on the vascular lumen. Areas of fibrinoid necrosis can be distinguished easily with the use of the PAS/Jones (silver) and the recognition of the granular texture rather than the glassy appearance of hyalin. The elastic lamina of the vessel wall as well as the interstitium between smooth muscle cells stain black from the silver. This finding is in sharp contrast to the bright pink staining of the fibrinoid material. Masson trichrome stain also delineates fibrinoid necrosis well as the fibrin stains bright red (Fig. 20.20). Thrombus formation is often superimposed on the area of necrosis, resulting in complete occlusion of the vessel. Fragmented red blood cells are frequently seen within either the thickened intima or the vessel wall, evidence of TMA (Fig. 20.21). Mixed inflammatory cells are occasionally present, but their presence is not necessary to make the diagnosis.
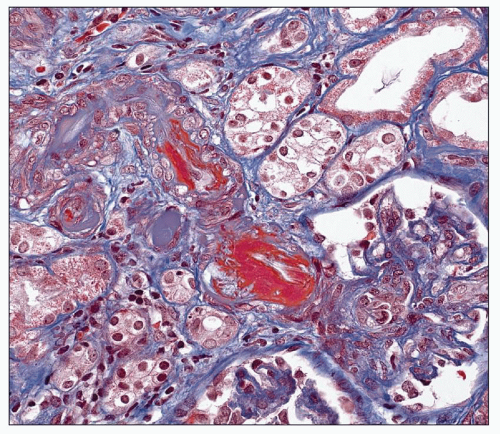 FIGURE 20.20 Afferent arteriole with fibrinoid necrosis staining bright red. The lumen is nearly totally occluded, resulting in collapse of the glomerulus being fed by the vessel. (Masson trichrome, ×310.) |
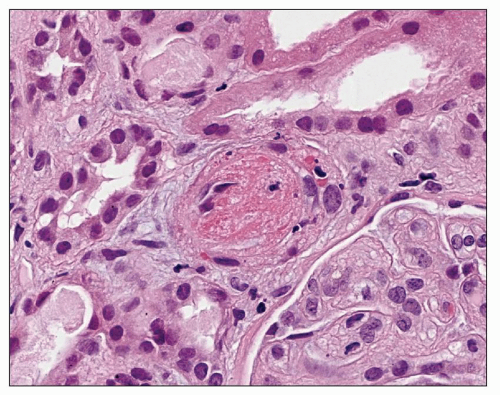 FIGURE 20.21 Arteriole showing TMA with platelet-fibrin thrombus occluding the lumen. Also note fragmented red blood cells within the vessel wall. (H&E, ×520.) |
Hughson et al. (132) in a study of patients with malignant nephrosclerosis identified nodules in small arteries and arterioles. These nodules were composed of spindled cells with a vascular network similar to the plexiform lesion seen in pulmonary hypertension. He hypothesized that the pathogenesis was due either to arterial necrosis or organization of thrombi or to a combination of the two, with the necrosis occurring first followed by the thrombus. Hughson also suggested that the intimal hyperplasia was due to turbulent blood flow. These changes were seen most commonly in the kidneys, followed by the periadrenal fat, pancreas, intestines, gallbladder, and heart.
THROMBOTIC MICROANGIOPATHY AND MALIGNANT NEPHROSCLEROSIS
The term malignant nephrosclerosis was first used by Fahr (133) in a description of the renal pathologic characteristics of a patient who had an accelerated or malignant form of hypertension. Bohle et al. (134) examined the issue of malignant nephrosclerosis in great detail. They divided their patients into two groups: one with the changes of fibrinoid necrosis in glomeruli and arterioles and the other with onionskin alterations in the intima of arteries. They designated the first group with either normal blood pressure or recent onset of hypertension as primary malignant hypertension. Bohle et al. believed that in these cases, the renal vascular morphologic changes preceded the hypertension. The other group had long-standing accelerated hypertension and was considered secondary malignant hypertension. The authors believed that the vascular changes in these cases were caused by the hypertension. Patients in the primary malignant hypertension group were younger than those in the secondary group. Furthermore, women outnumbered men, and the primary group had more severe hemolytic anemia and often presented in acute renal failure. The changes are those of TMA (discussed in Chapter 18) and are the same as seen in hemolytic-uremic syndrome,
thrombotic thrombocytopenic purpura, renal involvement in systemic sclerosis (scleroderma), toxicity of certain drugs such as cyclosporine and mitomycin C, and complications of radiation and bone marrow transplantation. It is characterized by fibrinoid changes in both glomeruli and renal arterioles accompanied by fragmented erythrocytes (see Fig. 20.20). The glomeruli may also show collapse and wrinkling of the tuft. The renal arterioles and small arteries show intimal thickening, often with a mucoid appearance resulting in marked narrowing of their lumina. As suggested in the name, thrombotic occlusion of the lumen is often superimposed on this narrowing.
thrombotic thrombocytopenic purpura, renal involvement in systemic sclerosis (scleroderma), toxicity of certain drugs such as cyclosporine and mitomycin C, and complications of radiation and bone marrow transplantation. It is characterized by fibrinoid changes in both glomeruli and renal arterioles accompanied by fragmented erythrocytes (see Fig. 20.20). The glomeruli may also show collapse and wrinkling of the tuft. The renal arterioles and small arteries show intimal thickening, often with a mucoid appearance resulting in marked narrowing of their lumina. As suggested in the name, thrombotic occlusion of the lumen is often superimposed on this narrowing.
The question of the primacy of malignant levels of hypertension in the pathogenesis of these microangiopathic lesions is still controversial. Most of the cases I see now that have the changes I consider representative of malignant nephrosclerosis are superimposed on the kidneys with primary glomerular disease. In these patients, severe renal parenchymal disease is present, and the history is consistent with preexistence of the glomerular disease before the onset of the malignant levels of hypertension. Thus, they can be considered as cases of secondary malignant hypertension. However, I have also seen a patient with IgA nephropathy and the changes of malignant nephrosclerosis in whom glomerular disease was not severe and the tubules and interstitium showed only focal fibrosis and atrophy. Nevertheless, the arteries showed changes consistent with the malignant hypertension, which was present clinically. In that case, I believe that the malignant hypertension was separable from the renal parenchymal disease and was thus primary or essential malignant hypertension. In most cases with microangiopathic changes, malignant hypertension is prominent clinically. However, I have seen the alterations of TMA without documentation of malignant levels of hypertension. In one particularly memorable case of scleroderma renal crisis, the patient never had a recorded blood pressure exceeding 120/80 mm Hg yet had spectacular fibrinoid necrosis of both glomeruli and arterioles.
Immunofluorescence Findings
Arterioles with hyaline accumulation have been shown to contain IgM and C3 most commonly, but IgG, IgA, IgE, and fibrinogen have also been recorded (135) (Fig. 20.22). C3 may also be observed without accompanying immunoglobulins. The most common reactant seen in immunofluorescence studies of malignant hypertension is fibrinogen (134). It may be noted in areas of fibrinoid necrosis, as would be expected, but it is also seen in glomerular capillary loops and in larger vessels without obvious fibrinoid necrosis. These changes are more prominent in the more active cases. On occasion, other immunoglobulins can be identified, presumably accumulating nonspecifically in areas of injury.
Electron Microscopy
Ultrastructural studies (122,136,137) of glomeruli from patients with hypertension demonstrated wrinkling and thickening of the GBM, which also becomes denser (Fig. 20.23). Electron microscopy confirms the presence of collagen fibrils with scattered cells within the capsular space and reduplication of the basement membrane material of the Bowman capsule (Fig. 20.24). The presumption has always been that the shrinkage of the tuft results from ischemia (136,137), but no good explanation has been advanced for the accumulation of collagenous material in the Bowman space. In open glomeruli, one may see mild but widespread subendothelial widening sometimes with new basement membrane formation (Fig. 20.25).
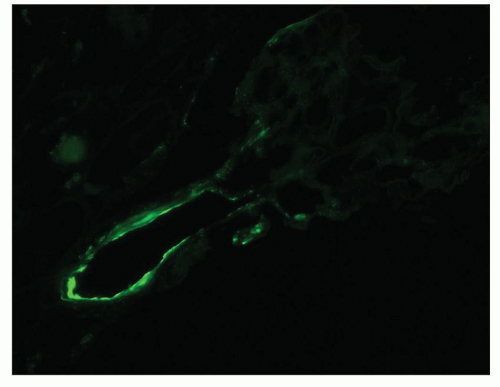 FIGURE 20.22 Immunofluorescence micrograph of afferent arteriole staining with C3 antiserum. (C3 antiserum, ×260.) |
Ultrastructural studies of intimal thickening of larger arteries in hypertension show that the intimal cells are myofibroblasts as characterized by both abundant rough endoplasmic reticulum and myofibrils distributed along the periphery of the cell. Immunohistochemistry with anti-smooth muscle-specific actin confirms the smooth muscle differentiation of these cells as well. Electron microscopy also demonstrates the presence of extracellular proteoglycans (small granules), which are responsible for imparting the mucoid appearance to the thickening (122,138).
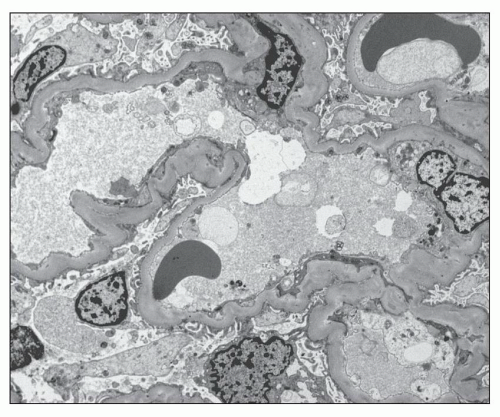 FIGURE 20.23 Electron micrograph showing wrinkling of the GBM with variability in its thickness. Foot processes are coarsened, but frank effacement is not seen. (Uranyl acetate and lead citrate, ×3600.) |
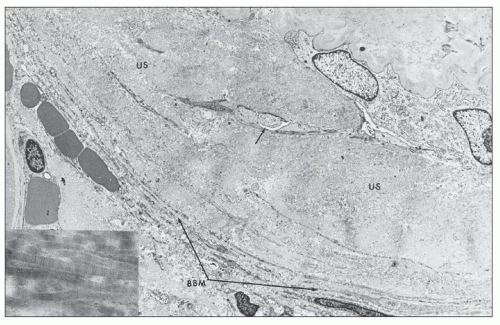 FIGURE 20.24 Electron micrograph from an obsolescent glomerulus. Remains of the capillary tuft with a wrinkled GBM and epithelial cell are seen in the top right corner. The urinary space (US) delimited by Bowman capsule basement membrane (BBM and arrows) is filled with collagen as confirmed at higher magnification in the lower left inset (×10,000; inset, ×36,000). (From Nagle RB, Kohnen PW, Bulger RE, et al. Ultrastructure of human renal obsolescent glomeruli. Lab Invest 1969;21:419. Copyright © 1969 by the U.S. Canadian Division of the International Academy of Pathology.) |
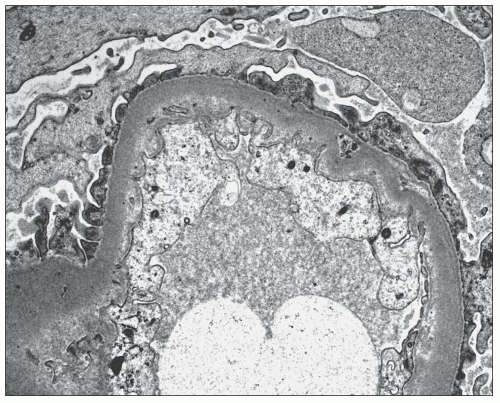 FIGURE 20.25 Electron micrograph showing mild subendothelial widening with new basement membrane formation. (Uranyl acetate and lead citrate, ×13,100.) |
Ultrastructural studies (122,139) of glomeruli from patients with accelerated hypertension demonstrate dense amorphous material in the areas of fibrinoid necrosis, sometimes with fibrin tactoids. The mesangium may be intact or show areas of loosening of the matrix, sometimes with finely granular material. Loss of attachment of the GBM to the matrix denotes areas of mesangiolysis (139), which results in capillary dilation and even microaneurysm formation, an alteration that can also be seen in the other conditions associated with TMA (see Chapter 18). Red blood cell fragments, platelets, and fibrin thrombi may be found in arterioles and capillaries. Endothelial cells may be swollen or, in more severely affected areas, they may become necrotic and disappear. The widened subendothelial space contains flocculent material of variable density, sometimes with fibrin tactoids. The glomeruli with long-term changes show wrinkling of the GBM, which becomes both thicker and denser. Areas with subendothelial widening have more flocculent material, which becomes more lucent, and new basement membrane material is laid down immediately below the endothelial cell (Fig. 20.26). Ultrastructural studies have suggested that the smooth muscle cells of the media are hypertrophied (122). Furthermore, such studies have also shown the presence of cells with smooth muscle features in the intima (122).
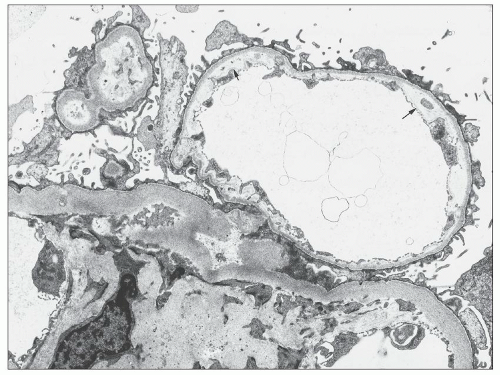 FIGURE 20.26 Electron micrograph of a portion of glomerular tuft showing a capillary loop with subendothelial widening and the new basement membrane (arrows) immediately subjacent to endothelial cell. The electron-lucent area between the two basement membranes contains flocculent material, most notably to the left. Occasional denser material may represent degraded fibrin-related material. (Uranyl acetate and lead citrate, ×5650.) |
Ultrastructural studies of vessels in severe hypertension confirm the presence of fibrin within fibrinoid necrosis, with frequent tactoids noted (122). Smooth muscle cells are necrotic or show various degenerative changes, including loss of microfibrils, vacuole formation, and increased numbers of autophagosomes. In those areas where the endothelium remains intact, one may see swelling of these cells or increased microfilaments, so-called stress fibers, at either the luminal surface or near its attachment to the basal lamina (140,141). An increase in the number of cellular organelles and myofilaments has been shown to occur in vascular endothelium when exposed to shear stress in vitro (142,143).
Etiology and Pathogenesis
Pathophysiology of Hypertension
The pathogenetic mechanisms of essential hypertension are complex, but it is recognized that sodium and fluid balance and vasomotor tone are the major determinants of blood pressure. A discussion of sodium and fluid balance is beyond the scope of this chapter. However, changes in vascular tone are related to structural changes in vessels. Thus, factors that affect vascular tone and autoregulation are described. Vascular resistance is determined by the Hagen-Poiseuille law and is directly related to the length of the vessels and the blood viscosity, as
represented by the hematocrit level. Resistance is inversely related to the fourth power of the diameter of the blood vessels (144). Arteriolar diameter is the most powerful determinant of resistance in the short term. In the long term, other factors, such as an increased wall-to-lumen ratio, which affects lumen diameter, and rarefaction, which results in a decrease in the number of arterioles and capillaries, also become important. Many factors contribute to the vascular changes including the sympathetic nervous system (SNS), the RAS, and other hormonal regulators of vascular tone, endothelial dysfunction, uric acid, and inflammation. These are discussed briefly in turn.
represented by the hematocrit level. Resistance is inversely related to the fourth power of the diameter of the blood vessels (144). Arteriolar diameter is the most powerful determinant of resistance in the short term. In the long term, other factors, such as an increased wall-to-lumen ratio, which affects lumen diameter, and rarefaction, which results in a decrease in the number of arterioles and capillaries, also become important. Many factors contribute to the vascular changes including the sympathetic nervous system (SNS), the RAS, and other hormonal regulators of vascular tone, endothelial dysfunction, uric acid, and inflammation. These are discussed briefly in turn.
The SNS is clearly involved in blood pressure regulation. Sympathetic activation occurs early in essential hypertension (145). Furthermore, the level of activation is directly related to severity of hypertension and is associated with greater cardiovascular morbidity and mortality (145). The SNS plays a role in chronic hypertension chiefly by increasing tubular sodium reabsorption, renin release, and renal vascular resistance (146). These changes shift the pressure-natriuresis curve to the right so that there is a resetting of arterial pressure up in order to reestablish sodium balance (146,147). SNS activity is increased in hypertensive patients even at an early stage of hypertensiveassociated renal dysfunction and increases with reduction of glomerular filtration rate (148). A number of mutations in genes associated with sympathetic activation have been recognized in essential hypertension as well as in experimental models of hypertension (61,145). The primary role of the SNS as a mechanism in the causation of hypertension is supported by the recent development of a technique to produce renal denervation by catheter-based radiofrequency ablation of renal nerves (149). Renal denervation was associated with long-term lowering of arterial pressure in a proof of principle study in patients with hypertension that had been difficult to control (149). The role of stress and the SNS in acute hypertension is well accepted (150). Mental stress has been shown to cause a larger increase in GFR in hypertensive patients than in normotensive subjects (146) perhaps indicating greater reactivity in vessels to SNS changes. Increases in SNS activity are also important in secondary hypertension.
The RAS plays a central role in blood pressure regulation. AII is the most potent of the peptides in the system with the AT1 receptor activating most hypertensive effects (147). AII causes renal vasoconstriction, increases the sensitivity of the tubuloglomerular feedback (TGF), and increases tubular reabsorption of sodium. AII may also have direct toxic effects on vessels and/or may be a stimulus to remodeling (see p. 867). Intrarenal AII levels are also increased in hypertension, resulting in the resetting of the pressure-natriuresis relationship. The classical view of the RAS in the kidney held that circulating AII exerted its effects on the afferent arterioles. Navar et al. (151) have studied the intratubular RAS and found abundant ATI receptors throughout the tubules of the nephron. Furthermore, intratubular concentrations of AII are higher than those of plasma, and AGT is synthesized in proximal tubule cells. Urinary AGT levels are elevated in hypertensive patients supporting the contention that this system is important in blood pressure regulation.
Endothelial dysfunction was initially defined as impaired vasodilation in response to such substances as acetylcholine or bradykinin. However, now it is recognized as a reduction in the expected vasoactive, anti-inflammatory, antiatherogenic, and anticoagulant properties of the endothelium induced by a reduction in NO bioactivity (152,153). Other factors involved in endothelial dysfunction include oxidative stress and up-regulation of inflammatory mediators. Endothelial dysfunction has been described in humans with hypertension and in families prior to the onset of hypertension (154). Some subsets of patients with hypertension do not show endothelial dysfunction (153). It is not yet clear whether endothelial dysfunction is a cause or effect in hypertension. However, its presence does signify increased risk for target organ damage.
NO is synthesized from L-arginine by a family of NO synthases and exists in two constitutive forms: endothelial and neuronal. The constitutive forms of NO are both calcium and calmodulin dependent. Release of the constitutive forms of NO produces various effects, including smooth muscle relaxation, inhibition of platelet aggregation and adhesion, and increased neurotransmission in peripheral sympathetic nerves. Larger, local doses affect both afferent and efferent arterioles. Loss or decrease in NO is one of the signs of endothelial dysfunction (152,153). Diminished NO can result from a decrease in endothelial NO synthase (eNOS) due to inhibitors or reduction in its substrate, arginine. NO synthase is inhibited by L-nitro ethyl arginine as well as by certain other substances that are normally produced endogenously (155). Long-term blockade of NO synthase brings about sustained hypertension and end-organ damage (156). RhoA, a member of the Rho family of small G proteins, plays a role in control of vascular tone (157). RhoA-Rho kinase signaling decreases eNOS synthesis by interfering with eNOS mRNA stability and possibly by stimulating arginase activity (157). Reactive oxygen species (ROS) have been associated with decreased NO mediated by an uncoupling of eNOS to a reductase function where it produces more ROS and less NO (158). Oxidative stress is defined by an imbalance between the concentration of free radicals and antioxidant defenses. During states of oxidative stress, ROS cause vasoconstriction and may be a precursor to hypertension. In the desoxycorticosterone-salt model of hypertension, Zhang et al. (159) showed that mitochondrial-related oxidative stress was associated with hypertension, which could be attenuated by inhibitors of the mitochondrial respiratory chain complex. In an in vitro model of vessels from skeletal muscle, Huang et al. (160) demonstrated that elevated shear stress could induce ROS that then diminished NO. Similarly, in human beings, markers of oxidative stress have been associated with lower levels of NO in hypertension and in chronic renal disease (152).
Hyperuricemia is also associated with increased blood pressure. Numerous epidemiologic papers including several longitudinal studies have established an associative risk between hyperuricemia and essential hypertension (161). This effect is more striking in young and middle-aged populations than in the elderly (96). Mazzali et al. (162) developed an animal model of hyperuricemia in the rat in which the hyperuricemia induced reduction in endothelial NO and stimulated renin production. Furthermore, they showed that the arteriolar lesions that developed were followed by the development of salt sensitivity that was independent of the level of serum uric acid (163). Additional studies showed that uric acid can enter vascular smooth muscle cells (VSMCs) and cause production of monocyte chemoattractant protein-1 (MCP-1) (164). Uric acid is also capable of up-regulating the production of CRP in human VSMCs (165), which leads us to a discussion of the role of inflammation in the pathogenesis of hypertension.
Numerous studies have demonstrated elevated circulating inflammatory markers in patients with hypertension (166,167). Increases in markers such as CRP or homocysteine, cytokines such as MCP-1, and adhesion molecules have been identified in patients with hypertension (167). These molecules are also elevated in prehypertensive states and indicate increased risk of developing hypertension (167). Homocysteine decreases NO bioavailability by enhancing lipid peroxidation evidenced by increased plasma levels of F2-isoprostanes (168). These isoprostanes stimulate production of ET-1, the most powerful vasoconstrictor known. ET-1 is produced by endothelial and vascular smooth muscle cells in response to such stimuli as shear stress, hypoxia, AII, and vasopressin. ET-1 interacts with ETA receptors to cause vascular smooth muscle contraction (169). Homocysteine and ET-1 have also been shown to have structural effects on vessels (168,170). AII also has proinflammatory actions.
Several authors in various reviews have suggested schemas that weave many of these strands into a common pathway to understand the pathogenesis of essential hypertension (161,166,171). It is possible that in the prehypertensive state repeated but intermittent episodes of vasoconstriction are initiated by a hyperactive SNS, by genetic alterations in the RAS, hyperuricemia, or a proinflammatory state. These various stimuli act through inducing oxidative stress, ET-1 release, and increased ROS to induce endothelial dysfunction and vasoconstriction. At first, the hypertension is salt resistant, and pressure natriuresis is intact. However, as arteriolar lesions develop due to effects from these various stimuli, ischemia results as well as defective autoregulation. Tubulointerstitial disease results, leading to increased sodium reabsorption. Another contributing factor to the impairment of autoregulation may be a reduction in nephron number (172,173). Arteriolar injury then reduces blood flow to glomeruli shifting the pressure-natriuresis curve and resulting in increased salt sensitivity and increased blood pressure.
The preceding discussion has introduced endothelial dysfunction and its attendant decreased NO induced by hyperuricemia, hyperhomocystinemia, and stimulated RAS as potential factors involved in the pathogenesis of hypertension. Advances in our knowledge of molecular biology have led to the uncovering of these substances and enhanced our understanding of their differing functions and interactions. In summary, the pathogenesis of hypertension remains a complex issue. Although we now can list many of the substances that play an important part in its genesis, we still do not understand all of the interrelationships among them.
Pathogenesis of Glomerular Lesions
EXPERIMENTAL STUDIES
The observations of Wilson and Byrom (174) using the 2K1C rat model, in which a renal artery from one kidney is clipped to reduce blood flow to induce hypertension, form the basis of much of our understanding of the pathologic manifestations of hypertension in the kidney. In this model, the clipped kidney is protected in the short term from hypertension, while the nonclipped kidney is exposed to the elevated systemic pressure and, as a result, manifests both glomerular and vascular injury. Changes in the nonprotected glomeruli include segmental or global necrosis with adhesions and eventual total sclerosis of the tuft. Fibrinoid necrosis is present in afferent arterioles and small renal arteries. These alterations are present in human malignant hypertension. Studies in various other experimental models of hypertension have confirmed these findings (175,176,177).
Many different experimental models of hypertension have been developed and are summarized by Dornas and Silva (17). Inhibition of the synthesis of NO synthase by nitro arginine methyl ester (NAME) produced vasoconstriction (156). When NO synthesis was inhibited for 2 months, blood pressure was elevated, the GFR decreased, proteinuria occurred, and glomerular injury appeared. Addition of sodium chloride to the diet exacerbated the hypertension and the glomerular injury (178). The glomerular lesions included glomerular collapse, segmental necrosis, and segmental glomerulosclerosis. Those glomeruli that showed collapse had decreased perfusion, whereas those glomeruli with either sclerosis or necrosis had been exposed to increased pressures.
Although ischemia due to vasoconstriction or luminal narrowing seems likely to be important in the collapsing form of glomerular injury in hypertension, it does not adequately explain all the alterations seen. Increased glomerular pressure may occur due to increased post-glomerular pressure, leading to increased pressure within the glomerulus itself. Early evidence that the direct transmission of increased pressure to the glomerulus could induce injury came from the studies of Byrom (179), in which removal of the clip from animals with 2K1C hypertension produced acute glomerular injury. More studies have supported the concept that increased glomerular pressure, whatever its pathogenesis, can be crucially important as a mechanism for direct glomerular injury. This increased glomerular capillary pressure results from the loss of effective afferent arteriolar constriction in the face of increased systemic pressure (i.e., loss of autoregulation) confirmed by micropuncture studies in various experimental models of hypertension (180,181,182). Autoregulation refers to the ability of the kidney to maintain renal blood flow and GFR in a narrow range despite wider variation in systemic blood pressure. This is accomplished chiefly through two mechanisms, namely, a myogenic reflex in the afferent arteriole and TGF (183). During impaired autoregulation, the sensitivity of TGF is reset due to changes in NO and AII. When there is sustained delivery of NaCl to the macula densa, the neuronal type of NO synthase is increased offsetting the usual vasoconstrictive effect of TGF on the afferent arteriole. These changes are found early in the course of the development of hypertension in the spontaneously hypertensive rat, the Milan hypertensive strain, and the Dahl salt-sensitive rat (reviewed in (183)). It is thought that this mechanism may be particularly important in salt-sensitive hypertension. The vasodilation resulting from the loss of autoregulation allows the transmission of higher pressure to the glomerulus, in effect an increase in glomerular capillary pressure.. The role of loss of autoregulation as one mechanism for the hemodynamic alterations has been studied extensively by Griffin et al. in several different models (184,185,186). In the normotensive renal ablation model, the systemic pressure is slightly higher than in controls but does not attain hypertensive levels. Nevertheless, when followed for 14 to 15 weeks, these animals develop glomerular lesions that correlate to the level of their systolic pressure as well as their pulse pressure (186). The authors believe that it is the transmission of this elevated pressure to the glomerulus that results in the glomerular injury. This supposition was also
supported by the comparison of changes in autoregulation in the stroke-prone spontaneously hypertensive rat (SHRsp) and their progenitors, the stroke-resistant SHR (184). Additional evidence for the role of loss of autoregulation in glomerular injury in the setting of hypertension was provided in a comparison of four different models of hypertension (177). The two models with increased preglomerular resistance manifest less glomerular injury than the two models with decreased preglomerular resistance (177). Glomeruli throughout the cortex may be affected, with juxtamedullary glomeruli affected slightly more frequently (177). In models with vasoconstriction, the glomerular injury is much less severe and is almost restricted to juxtamedullary glomeruli (177). This finding is thought to be related to a greater loss of autoregulation in juxtamedullary as compared with superficial glomeruli in these two models.
supported by the comparison of changes in autoregulation in the stroke-prone spontaneously hypertensive rat (SHRsp) and their progenitors, the stroke-resistant SHR (184). Additional evidence for the role of loss of autoregulation in glomerular injury in the setting of hypertension was provided in a comparison of four different models of hypertension (177). The two models with increased preglomerular resistance manifest less glomerular injury than the two models with decreased preglomerular resistance (177). Glomeruli throughout the cortex may be affected, with juxtamedullary glomeruli affected slightly more frequently (177). In models with vasoconstriction, the glomerular injury is much less severe and is almost restricted to juxtamedullary glomeruli (177). This finding is thought to be related to a greater loss of autoregulation in juxtamedullary as compared with superficial glomeruli in these two models.
The endothelial injury from this increased glomerular pressure may be due to shear stress, direct mechanical injury, and/or oxidative stress. The forces that bring about visceral epithelial cell injury have been studied by Kretzler et al. (187) and involve capillary loop distension with epithelial cell hypertrophy and eventual loss of attachment of the epithelial cell to the GBM. In addition, epithelial cells may show blebs and vacuoles. A possible link between mechanical strain and podocyte injury has been studied in vitro by Durvasula et al. (188). They found that increased mechanical strain was followed by increased AII production, a fivefold increase in AT1 receptor mRNA expression, and an increase in transforming growth factor-beta (TGFβ) mRNA expression. This was also accompanied by an increase in apoptosis of the epithelial cells (188). Endothelial dysfunction may precede hypertension and is associated with decreased NO levels and an increase in AII resulting in impaired vasodilation, platelet activation, and increased vascular permeability (189,190). Microalbuminuria is a marker of endothelial dysfunction. The Ren-2 transgenic rat model of hypertension has increased tissue activity of the RAS and manifests hypertension, proteinuria, and insulin resistance (191). Effacement of foot processes is noted in glomeruli of such rats accompanied by decreased cortical expression of nephrin (191). Increased levels of 3-nitrotyrosine (3-NT), a surrogate for lipid peroxidative and oxidative stress, were present in renal cortex of Ren-2 rats. Treatment with a specific renin inhibitor restored the nephrin expression and the foot processes. Concomitant decrease in 3-NT was also found (191). In previous experiments, these investigators had shown that Nebivolol, a beta blocker that increases NO synthesis by endothelial cells, abrogated the proteinuria in Ren-2 rats accompanied by decrease in ROS/NADPH oxidase production (192).
CLINICAL STUDIES
Obsolete glomeruli show two different forms. In the first of these forms, the obsolescent glomeruli are characterized by wrinkling of the GBM with retraction of the tuft to one side of the Bowman capsule and collagenization of the Bowman space (see Fig. 20.5). This may be viewed as a form of collapse of the glomerular tuft. In the second form, the glomeruli are solidified or hyalinized; thus, an increase in matrix is often accompanied by remnants of hyalinosis lesions (Fig. 20.27). The obsolescent lesion is predominant in essential hypertension. The wrinkled glomerulus is likely the result of chronic ischemia. Kasiske (193) compared autopsy kidneys from patients with moderate to severe systemic atherosclerosis to those from patients of similar age with mild atherosclerosis with respect to the number of sclerotic glomeruli, the degree of vascular narrowing of arcuate and interlobular arteries, and the presence or absence of hypertension. He found that the extent of glomerular obsolescence depended on both the age of the patient and the degree of intrarenal vascular disease. Although hypertension correlated with the degree of glomerulosclerosis, it was not independent of the other two factors. The degree of narrowing of the renal artery correlated strongly with the extent of glomerular obsolescence, a finding supporting the idea that the sclerosis was due to ischemia, as represented by the vascular narrowing. It is well known that glomeruli may become sclerotic with increasing age, independent of increased blood pressure (110,194). Kaplan et al. (110) stated that 95% of the population aged ≤40 years should have fewer than 10% globally sclerotic glomeruli. Kappel and Olsen (194) countered that no more than 1% of glomeruli should be obsolete by age 40 but that as many 30% of glomeruli may become globally sclerotic by age 80.
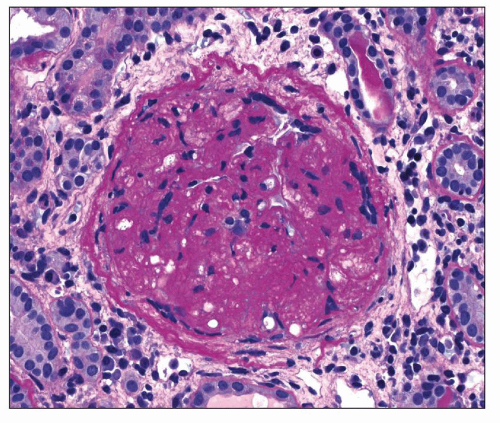 FIGURE 20.27 Solidified glomerulus extending to the Bowman capsule with increased mesangium, sclerosis of capillary loops, and occasional foam cells. (PAS, ×400.) |
Hypoxia has been heavily implicated in the pathogenesis of interstitial fibrosis (126,195) (see p. 870). Recent studies suggest possible mechanisms for a role in glomerulosclerosis as well. Neusser et al. (196) examined gene expression data in microdissected glomeruli from patients with hypertensive nephropathy and found an increased expression in several target genes of hypoxia-inducible factor (HIF). Furthermore, HIF-1α was identified in podocyte nuclei as a marker of transcriptional activation.
The solidified glomerulus is the alteration that Bohle described in DBN (113,114), a form associated with a more rapid course to ESRD. Marcantoni et al. (74) studied 62 hypertensive patients (19 African Americans and 43 Caucasians) with regard to the types of glomerular injury present in their renal biopsies. They found that 25% of glomeruli in the African American patients were of the solidified form as compared to 8% in Caucasians. A subgroup of patients also had segmental sclerosis, also seen more frequently in African Americans. In
addition to the segmental lesions, the African Americans in this subgroup had 38% solidified glomeruli. The higher prevalence of the solidified glomerulus in African Americans with hypertension was first associated with the more common occurrence of polymorphisms in the nonmuscle myosin heavy chain 9 (MYH9) gene in African Americans (69). Polymorphisms in MYH9 have been linked to FSGS and other renal diseases in both African Americans and Caucasians (69). More recently, this association has been found to be due to missense mutations in apolipoprotein-1, which is closely linked to MYH9 (76). Loss of autoregulation has also been suggested as a potential cause of the solidified glomerulus (111,116,197). Hill et al. (116) studied the relationship between arteriolar diameter and glomerular appearance in hypertensive individuals and found that no correlation was present in normal appearing glomeruli. However, hypertrophied glomeruli that were solidified or showed FSGS-like lesions demonstrated a correlation between arteriolar diameter and glomerular size. They suggested that this finding supported loss of autoregulation (116). Another factor in the pathogenesis of these lesions may be the presence of reduced numbers of glomeruli in some patients with hypertension (8,172,173). Brenner and Chertow (172) suggested that early gestational age or fetal growth retardation might affect nephrogenesis so that a reduced number of nephrons occur with consequent hyperfiltration of those that are present. Keller et al. (173) found a reduced number of nephrons at autopsy in patients with hypertension. Podocyte loss may also contribute to this lesion. Wang et al. (118) have demonstrated decreased podocyte number in glomeruli from hypertensive patients as compared to glomeruli from donor kidney biopsies. Podocyte loss has been recognized as a risk factor in the development of FSGS as well as other glomerular lesions (198).
addition to the segmental lesions, the African Americans in this subgroup had 38% solidified glomeruli. The higher prevalence of the solidified glomerulus in African Americans with hypertension was first associated with the more common occurrence of polymorphisms in the nonmuscle myosin heavy chain 9 (MYH9) gene in African Americans (69). Polymorphisms in MYH9 have been linked to FSGS and other renal diseases in both African Americans and Caucasians (69). More recently, this association has been found to be due to missense mutations in apolipoprotein-1, which is closely linked to MYH9 (76). Loss of autoregulation has also been suggested as a potential cause of the solidified glomerulus (111,116,197). Hill et al. (116) studied the relationship between arteriolar diameter and glomerular appearance in hypertensive individuals and found that no correlation was present in normal appearing glomeruli. However, hypertrophied glomeruli that were solidified or showed FSGS-like lesions demonstrated a correlation between arteriolar diameter and glomerular size. They suggested that this finding supported loss of autoregulation (116). Another factor in the pathogenesis of these lesions may be the presence of reduced numbers of glomeruli in some patients with hypertension (8,172,173). Brenner and Chertow (172) suggested that early gestational age or fetal growth retardation might affect nephrogenesis so that a reduced number of nephrons occur with consequent hyperfiltration of those that are present. Keller et al. (173) found a reduced number of nephrons at autopsy in patients with hypertension. Podocyte loss may also contribute to this lesion. Wang et al. (118) have demonstrated decreased podocyte number in glomeruli from hypertensive patients as compared to glomeruli from donor kidney biopsies. Podocyte loss has been recognized as a risk factor in the development of FSGS as well as other glomerular lesions (198).
Pathogenesis of Small Vessel Changes
Considerable examination of the pathogenesis of the different vascular changes seen in human hypertension has been undertaken over the years. These changes include hyaline arteriolosclerosis, intimal thickening, and medial thickening. Each of the major alterations is discussed in turn.
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Glomerular Diseases With Organized Deposits
Glomerular Diseases With Organized Deposits
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


