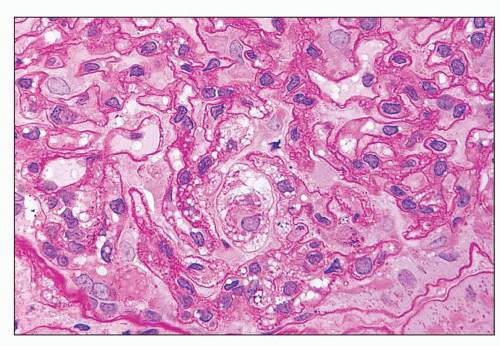
 FIGURE 27.1 Glomerulus from a patient with LCAT deficiency. It shows mesangial and focal capillary wall thickening with prominent bubbly lipid interposed between what appear to be two layers of basement membrane (double contours). (Periodic acid-Schiff [PAS]; ×400.) |
and mesangial deposits (8); these observations contrast with sequential assessment in a kidney allograft where deposits were initially identified in the mesangium (9). The lipid deposits are partly lucent and partly deeply osmiophilic, the latter including cross-striated curvilinear serpiginous fibrils, rounded lamellar densities, and granular densities (5,6). The former two are predominantly in epimembranous and intramembranous deposits, while granular densities are predominantly in subendothelial deposits. Densely osmiophilic basement membrane deposits have resembled the glomerular alterations of densedeposit disease and may increase basement membrane fragility because focal disruptions are identifiable (10). Mesangial deposits tend to be large and dense, comprising increased matrix and hyaline. Foam cells may be present in the mesangium, as shown by light microscopy; they rarely seem to be endocapillary. Arteriolar endothelial and medial cells may also contain lipid deposits (5). Tubular atrophy and interstitial fibrosis progress variably.
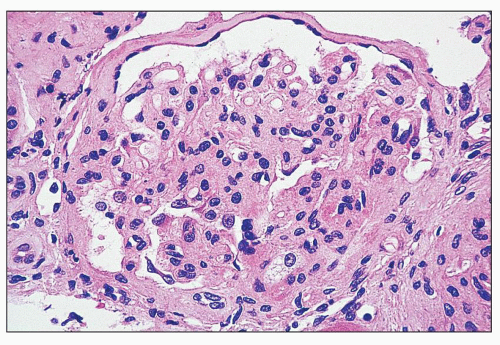 FIGURE 27.2 Glomerulus in LCAT deficiency showing thickened basement membranes and mesangial foam cells (bottom) entrapped within increased eosinophilic matrix. (Hematoxylin & eosin [H&E]; ×200.) |
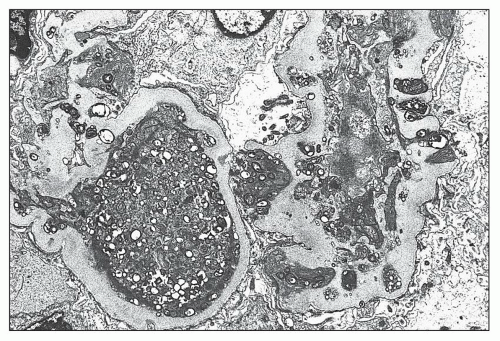 FIGURE 27.3 Electron microscopy in LCAT deficiency shows glomerular epimembranous, intramembranous, subendothelial, and mesangial accumulations of extracellular lipid material with membranous profiles and granules. (×9000.) |
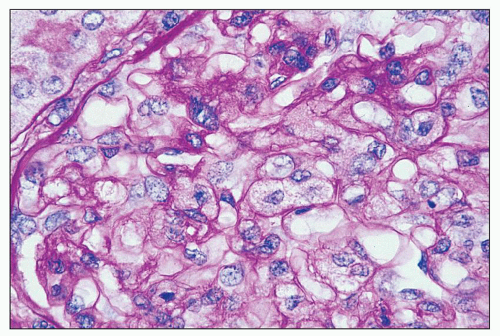 FIGURE 27.4 Glomerulus from renal allograft in a patient with LCAT deficiency, 7 weeks after transplantation, with recurrence of foamy mesangial cells. (PAS; ×400.) |
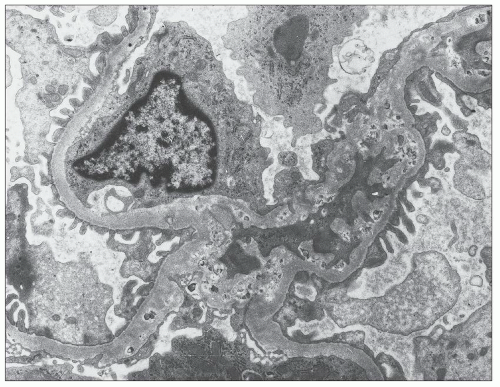 FIGURE 27.5 Electron microscopy shows recurrence of mesangial and subendothelial lipid deposits in renal allograft at 7 weeks after renal transplantation in a patient with LCAT deficiency. (×10,400.) |
of diabetic nephropathy, the incidence, severity, and response to therapy of various other nephropathies, and the serum lipid profile and the risk of atherosclerosis in end-stage renal disease (ESRD) (30). Novel missense mutations and deletions in the ApoE gene are thought to be pathogenic in LPG, and their existence was proven by sequencing after observing discordant results between restriction fragment genotyping and ApoE phenotyping via isoelectric focusing. A growing number of mutant isoforms—ApoE Sendai, Kyoto, Tokyo/Maebashi, Tsukuba, Chicago, Okayama, Guangzhou, Hong Kong, Modena, and Las Vegas (each genotype named after the index patient’s city of origin) and ApoE1 and ApoE5—have
been associated with LPG (31), and the development of LPG in ApoE Sendai-infected mice suggests an etiologic role for these atypical isoforms (32,33). Some of the mutations can alter the tertiary structure of the variant apolipoprotein, thereby affecting interactions with receptors and cell surfaces that may induce abnormal intraglomerular lipid trafficking (34,35). Multiple family members may carry the same ApoE mutation in a heterozygous form, but not all family members with mutant isoforms develop LPG. This observation implies that LPG is a dominantly inherited disease with incomplete penetrance and that other genetic or environmental factors participate in its pathogenesis. For example, alterations in lipoprotein metabolism may be detrimental. The Fc receptor γ on macrophages and mesangial cells is involved in the recognition and clearance of LDL. The development of LPG in Fc receptor γ-deficient mice suggests that a reduction in LDL clearance may induce lipoprotein deposition (36). As lesions are localized to the glomeruli, the role of intrinsic mesangial cells seems paramount. The exact mechanism of the renal disease, however, remains to be defined, but variant ApoE appears to be a prerequisite. With one exception, all patients described thus far were found to be heterozygous for ApoE gene mutations (37).
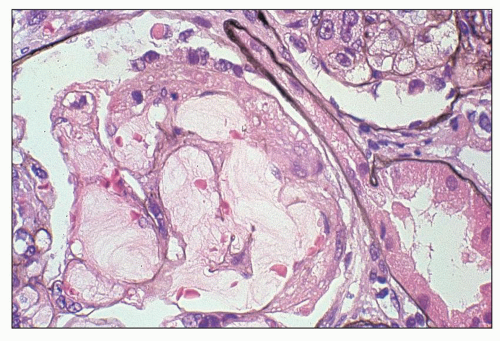 FIGURE 27.6 The glomerulus from a patient with lipoprotein glomerulopathy. The capillaries are distended by pale lipoprotein thrombi that have a vague laminated appearance. Dilation of the capillary is associated with mesangiolysis. (PAS-silver methenamine; ×200.) |
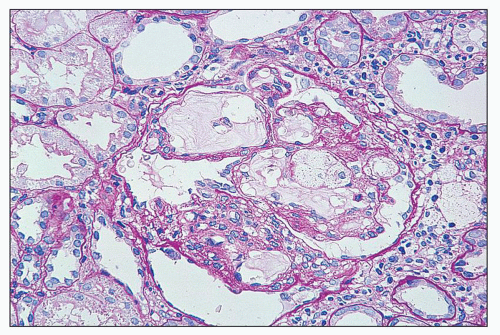 FIGURE 27.7 Capillaries in the glomerulus with lipoprotein glomerulopathy are distended with lipoprotein thrombi. The lobule at the bottom has increased mesangial cellularity and matrix. There are multiple adhesions to Bowman capsule, which will progress with sclerosis and hyalinosis. (PAS; ×100.) |
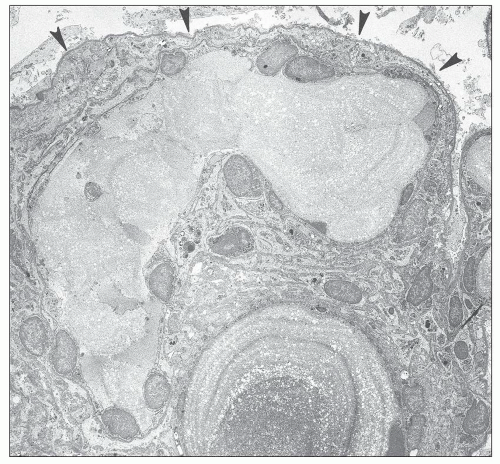 FIGURE 27.8 Electron micrograph shows the glomerular capillary lumina to be filled with partially lamellated, finely vacuolated lipoprotein thrombi. The mesangium is thickened by cell processes and increased matrix. The capillary wall is also thickened, with mesangial interposition and duplication of the glomerular basement membrane (arrowheads). (×1900.) |
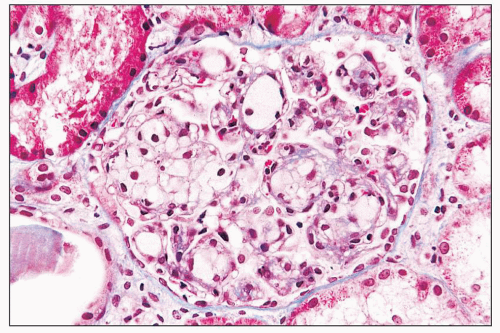 FIGURE 27.9 Glomerulus from a child with type III hyperlipoproteinemia that shows groups of foam cells in the mesangium and distending the capillary lumens. (Masson trichrome; ×400.) |
Gaucher disease, the most common lysosomal storage disease, only becomes symptomatic after splenectomy, a therapeutic procedure in type 1 nonneuropathic disease that is performed less often in the era of enzyme replacement (51,52,53). Its characteristic “wrinkled paper” appearance allows the diagnosis of Gaucher disease by light microscopy, whereas the pathology of the majority of lysosomal disorders is not distinct. Rather, storage cells are typified by clear and sometimes foamy cytoplasm, the consequence of storage material dissolving with processing (54). Special histochemical staining, especially on frozen sections where water and alcohol-soluble substances are preserved, may help to characterize the stored material, but is frequently nonspecific. Electron microscopy can usually define the disorder further. In most instances, the diagnosis will already be known from the history, laboratory data, and enzyme analysis.
normal estimated glomerular filtration rate (GFR), without manifestation of overt proteinuria or even microalbuminuria (74,75,76). As clinical parameters lack sensitivity, kidney biopsy has been employed by some to assess baseline injury (74,76). Proteinuria seems to be the most important predictor for renal progression, especially for men, in whom advanced nephropathy is more prevalent and generally occurs earlier (77,78). Most females have slowly progressive kidney disease; however, the subset (15% to 20%) that develops ESRD does so at approximately the same age as men (79). Loss of urine-concentrating ability leads to polyuria and polydipsia, and altered tubular functions have also been identified, such as impaired glucose and potassium reabsorption, to a greater degree than can be accounted for by reduced GFR (80). The urine sediment contains lipid globules showing Maltese crosses on polarization and desquamated cells containing myeloid bodies (81).
TABLE 27.1 Renal involvement by lysosomal storage disorders (without significant functional impairment) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
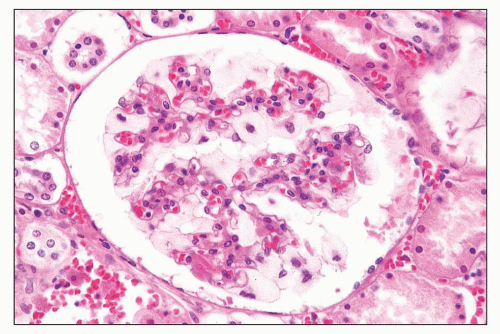 FIGURE 27.10 Glomerulus in I-cell disease has profoundly enlarged, finely vacuolated podocytes. (H&E; ×400.) |
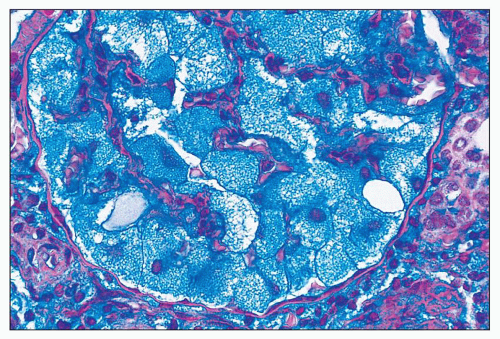 FIGURE 27.11 The vacuolated podocytes in I-cell disease contain abundant glycolipids and acidic glycosaminoglycans. (Hale colloidal iron; ×400.) |
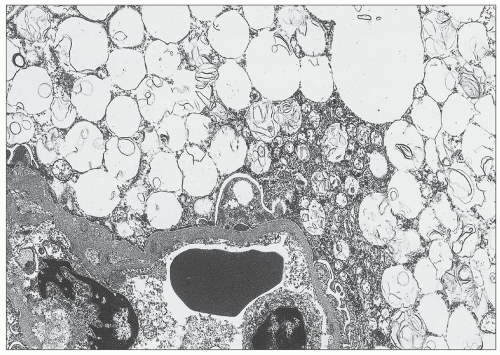 FIGURE 27.12 Electron micrograph of a podocyte in I-cell disease shows that the vacuoles contain a few membranous and lamellated inclusions. The material in the largely “empty” vacuoles may have been dissolved during processing. (×9215.) |
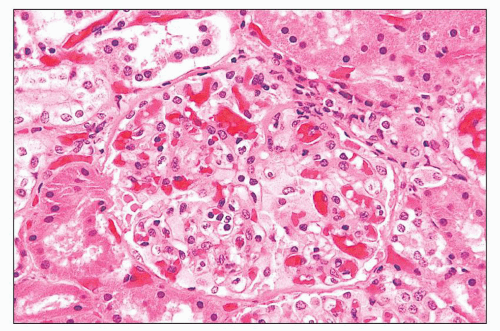 FIGURE 27.13 Glomerulus from a 13-year-old boy with neuronal ceroid lipofuscinosis. The child had severe cerebral atrophy and neurologic impairment but normal renal function. The visceral podocytes are distended with granular ceroid material. (H&E; ×200.) |
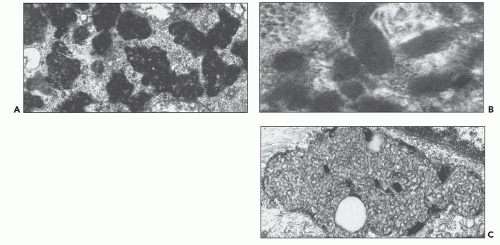 FIGURE 27.14 Electron microscopy of storage material in neuronal ceroid lipofuscinosis showing characteristic granular osmiophilic bodies (A) and fingerprint (B) and curvilinear (C) profiles. (×31,500.) |
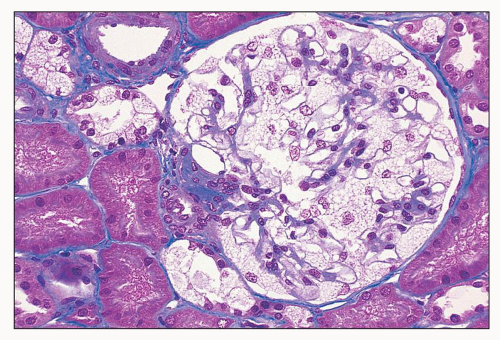 FIGURE 27.15 The glomerular podocytes are swollen and finely vacuolated in a patient with Fabry disease. Epithelial cells of distal tubules are also vacuolated. (Mallory trichrome; ×200.) |
or formalin fixed, is birefringent, autofluorescent, sudanophilic, and positive to oil red O and PAS. It may also be demonstrated in frozen sections by lectin binding (85). A similar vacuolated appearance, variable in quantity but sometimes considerable, is present in tubular epithelial cells, particularly of the distal tubules and the loop of Henle (Figs. 27.15 and 27.17). Small arteries and arterioles show vacuolation of the endothelial cells and finely vacuolated areas in the smooth muscle (Fig. 27.18). Interstitial foam cells can be seen. Progression of the disease leads to mildly increased mesangial matrix and cellularity with segmental glomerular sclerosis (Fig. 27.19), capillary wall thickening, tubular atrophy, interstitial fibrosis, and arterial and arteriolar sclerosis. Immunofluorescence is negative or nonspecific. Storage of myeloid bodies has been shown also in the liver and spleen (86). Hemizygotes have more severe lipid storage than do heterozygotes (75,87,88).
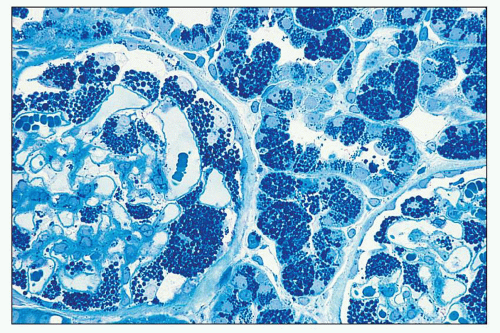 FIGURE 27.16 The intracellular lipid inclusions in Fabry disease are preserved in osmicated, epoxy-embedded tissue. The enlarged podocytes and tubular epithelial cells contain lamellated inclusion bodies (same patient as in Fig. 27.15). (Methylene blue; ×250.) |
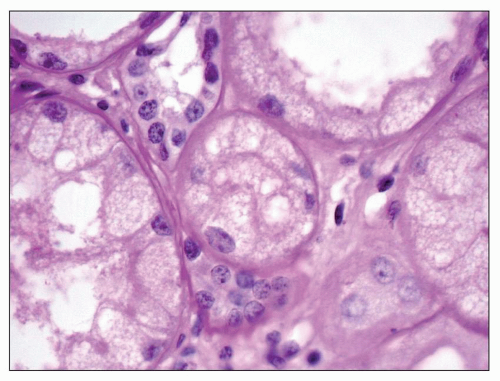 FIGURE 27.17 Fine, foamy vacuolation of tubular cells from a patient with Fabry disease. (PAS; ×400.) |
Recently described are unique subendothelial deposits associated with basement membrane duplication and composed of membrane-like material arranged in geographic layers (88).
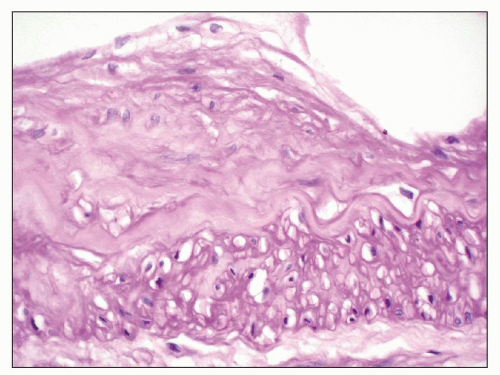 FIGURE 27.18 Renal artery in end-stage Fabry disease has moderate intimal fibroplasia, cleared endothelial cells (top), and empty spaces in the media (bottom). (PAS; ×400.) |
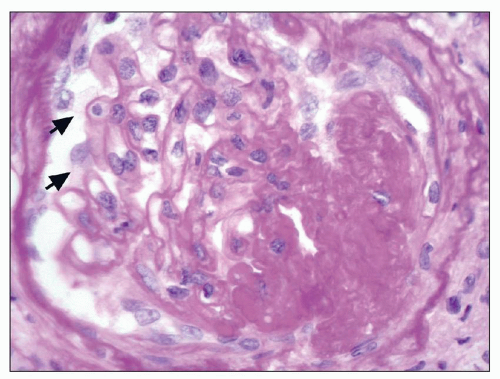 FIGURE 27.19 Glomerulus in Fabry disease shows thickened capillary walls and partial solidification. Fine vacuolation is still evident in a few podocytes (arrows) over an intact portion of the tuft. (PAS; ×400.) |
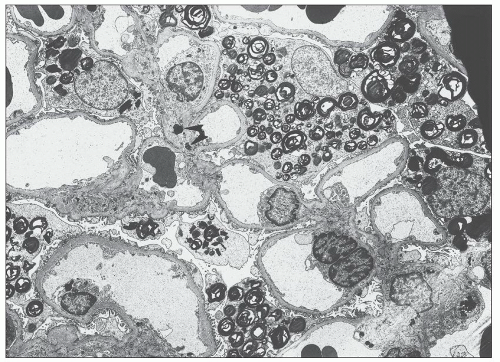 FIGURE 27.20 Electron micrograph of glomerulus in Fabry disease shows lamellated lipid inclusions (“myeloid bodies”) in podocytes. A few mesangial inclusions are also present (arrowheads, just above the center). (×3000.) |
Stabilization of kidney functional decline has been shown, with consistently better results when therapy is initiated before evidence of significant proteinuria (107,108). Recent efforts are focused on the addition of renin-angiotensin system blockade to augment effectiveness (104). Enzyme replacement therapy in kidney transplant patients with Fabry disease also appears to be safe and often effective against extrarenal involvement (100,106).
although they stain lightly with colloidal iron, indicating partial preservation of the material during processing. PAS staining shows only a fine granularity. Tubular cells, especially in proximal tubules, and interstitial cells are also vacuolated. Cytoplasmic vacuoles have also been found in endothelial cells of renal vessels (123).
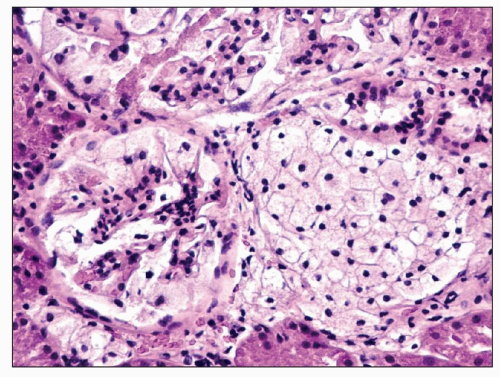 FIGURE 27.21 The glomerulus in nephrosialidosis contains vacuolated podocytes that fill Bowman space. The adjacent interstitium and tubules contain vacuolated storage cells. (H&E; ×200.) |
CblC defect can be difficult to diagnose on a clinical basis due to its heterogeneous manifestation but is established by finding the combination of methylmalonic aciduria, homocystinuria, and increased propionylcarnitine with normal serum vitamin B12 and transcobalamin II concentrations.
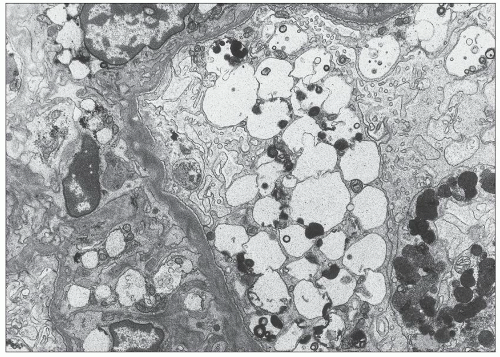 FIGURE 27.22 Electron microscopy of a glomerulus in nephrosialidosis shows vacuolated podocytes and mesangial cells. The vacuoles are partially filled with electron-dense bodies and also contain lucent and finely granular material. (×10,400.) (Courtesy of Drs. C. E. Kashtan and Z. Posalaky.) |
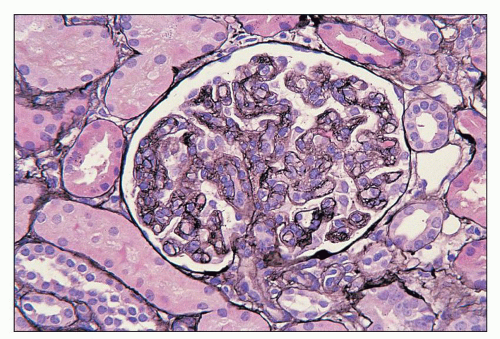 FIGURE 27.23 Mildly hypercellular glomerulus from a 5-year-old boy with cobalamin C deficiency. Chronic microangiopathic changes include thickened and duplicated basement membranes without capillary thrombi. (PAS-silver methenamine; ×400.) |
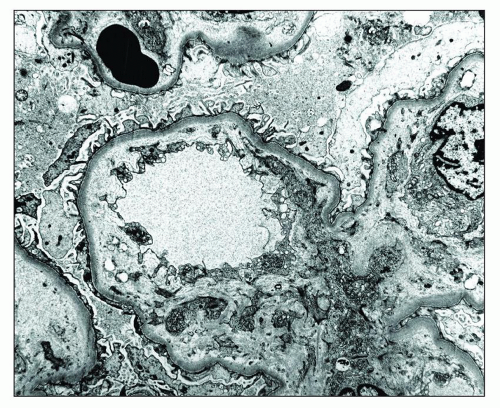 FIGURE 27.24 Electron microscopy in cobalamin C deficiency showing narrowing of the capillary lumen by subendothelial granular and fibrillar material and focal mesangial cell interposition. Foot process fusion is only focal. (×2500.) |
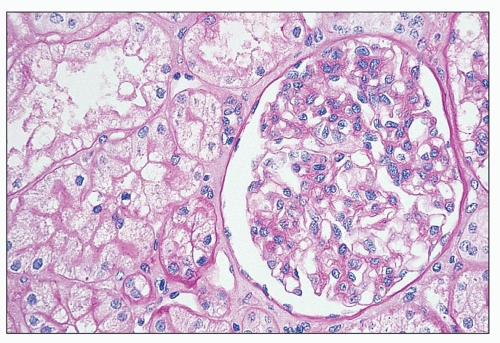 FIGURE 27.25 A glomerulus in a patient with GSD-I is hypertrophied and has mildly increased numbers of prominent mesangial cells. Adjacent tubules show intense vacuolation of epithelial cells. (PAS; ×100.) |
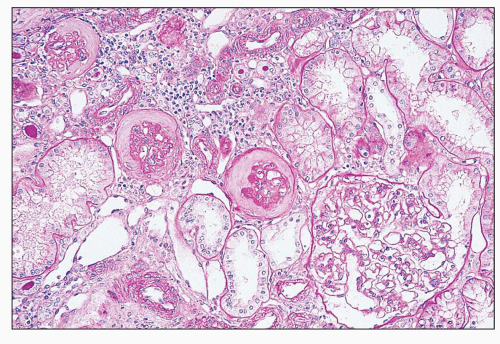 FIGURE 27.26 Nephrectomy kidney from a child with GSD-I shows an enlarged glomerulus and extensive global glomerulosclerosis. Atrophic tubules are associated with interstitial inflammation, fibrosis, and thickened arteries. (PAS; ×50.) |
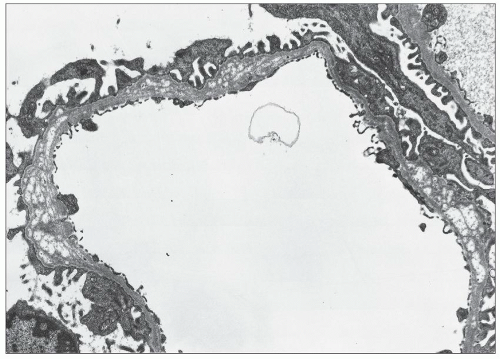 FIGURE 27.27 Electron microscopy shows the glomerular basement membrane in GSD-I to be frequently lamellated, incorporating irregular lucencies and fine granules. (×12,000.) (Courtesy of Dr. R. Verani.) |
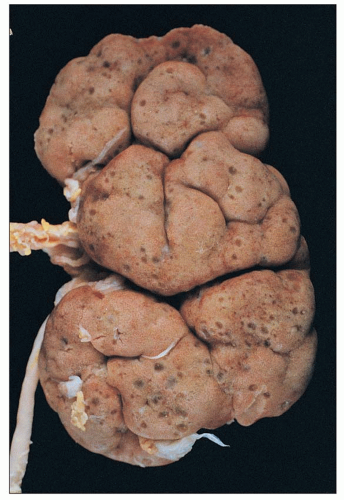 FIGURE 27.28 Autopsy kidney from an infant with Zellweger syndrome shows prominent fetal lobulation and numerous small, thin-walled cysts in the peripheral cortex and subcapsular area. |
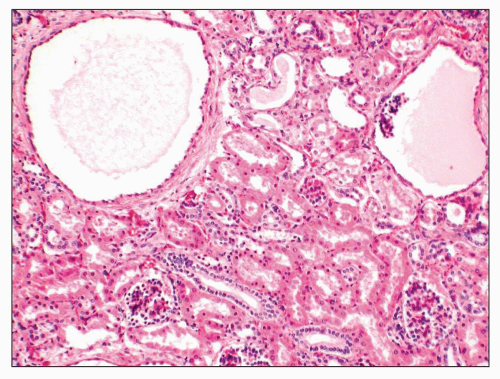 FIGURE 27.29 Microcysts of both tubular and glomerular origin are evident in the cortex of a Zellweger kidney without significant functional implication. (H&E; ×100.) |
become evident. High concentrations of protein are present in the spinal fluid. Full expression of the disease occurs during the fourth or fifth decade, but it can manifest in childhood.
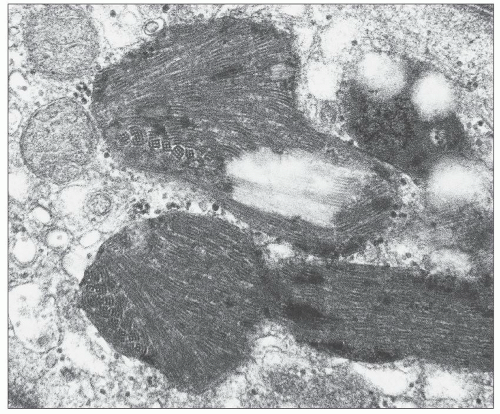 FIGURE 27.30 The tubular epithelial cells in adult Refsum disease contain crystalloid inclusions, with geometric structures. (×77,000.) (Courtesy of Dr. B. Panner.) |
recognized: (a) infantile oxalosis with early nephrocalcinosis and kidney failure; (b) childhood recurrent urolithiasis and rapidly progressive renal failure; (c) late onset with only occasional stone passage in adulthood; (d) post-kidney transplantation recurrence; and (d) presymptomatic discovery with family screening (176). Approximately 10% of patients have severe disease, with early infantile onset manifesting as failure to thrive, severe metabolic acidosis, anemia, and rapid progression to renal failure, whereas another 10% may not become symptomatic until the fourth or fifth decades (177). Progressive parenchymal deposition of calcium oxalate impairs renal function, which ultimately leads to systemic oxalosis. Complications include severe deforming osteopathy, arthropathy, cardiomyopathy, retinopathy, neuropathy, and pancytopenia. The kidneys are often small and may feel gritty on cut section (Fig. 27.31). Small, polyhedral or rhomboid, usually transparent, doubly refractile crystals are recognized histologically and accumulate in tubules, where they compress and destroy epithelium. The crystals can extend into the interstitium and induce fibrosis (Fig. 27.32). End-stage kidneys show extensive glomerulosclerosis and widespread interstitial fibrosis that encases abundant crystals (Fig. 27.33). Stone analysis demonstrates virtually pure (>95%) calcium oxalate monohydrate (whewellite) and a whitish or pale-yellow surface with a loose, unorganized center comprised of spherical, variably sized crystal aggregates, approximately 50 µm in diameter, that resemble balls of wool (178).
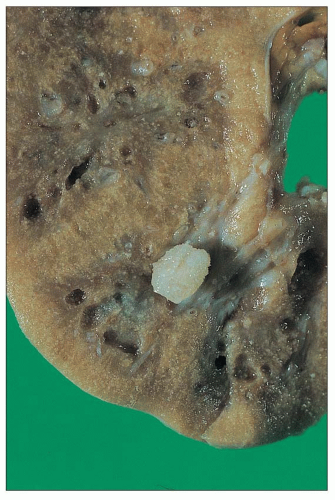 FIGURE 27.31 Autopsy kidney of 5-year-old boy with primary hyperoxaluria who presented at 5 months with seizures and failure to thrive. The kidneys were one third the expected weight and had a gritty consistency caused by yellow-tan oxalate crystals. A 0.3-cm calculus occupies a calyx. (Courtesy of J. Siebert, Ph.D., Seattle Children’s Hospital, University of Washington.) |
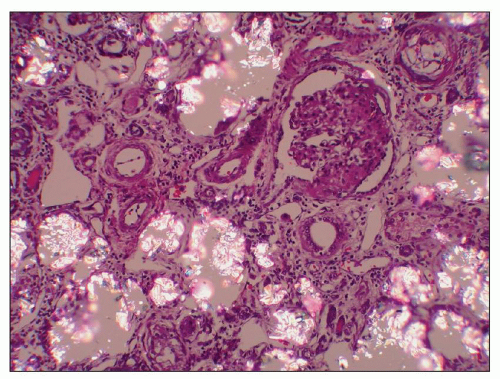 FIGURE 27.32 Renal tubules in primary hyperoxaluria are filled with rhomboid and polyhedral refractile oxalate crystals. A glomerulus is collapsed and segmentally sclerotic (same kidney as Fig. 27.31). (H&E, partial polarization; ×200.) |
levels of urinary oxalate usually indicate PH in the absence of any likely causes of secondary hyperoxaluria, either increased intake (excessive star fruit and peanut ingestion), increased absorption (“enteric hyperoxaluria” related to orlistat therapy or gastrointestinal disease or surgery), or increased production (ascorbic acid or ethylene glycol ingestion). Histologic demonstration of calcium oxalate deposition in the kidney has been used for diagnosis, but it is not specific for primary disease; determining the AGT activity in a liver biopsy sample has been considered the gold standard for diagnosis. Molecular genetics has now reached a level of sensitivity and specificity that makes it useful for definitive testing and is also considered the preferred method for prenatal testing (179). More than 150 mutations have been identified in the AGXT gene, which resides on chromosome 2q37.3 (180,181). Mutations result in accelerated proteolysis, peroxisome-to-mitochondrion targeting defects, intraperoxisomal AGT aggregation, absence of AGT catalytic activity, and absence of both catalytic activity and immunoreactivity. The clinical heterogeneity may relate to great variability in enzymatic activity among patients but is clearly influenced by potential modifier genes, environmental factors, and genetic background, as the genotype-phenotype correlation is limited. However, some mutations that result in AGT mistargeting appear to be associated with responsiveness to pyridoxine treatment (181,182,183,184,185,186).
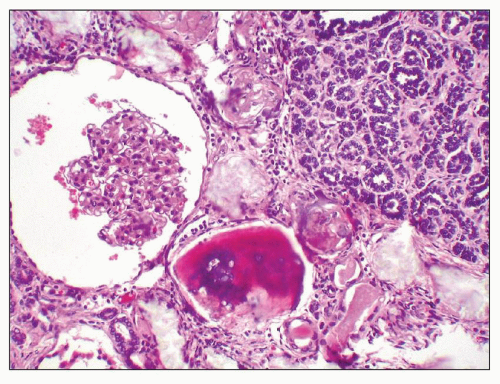 FIGURE 27.33 The end-stage kidney in primary hyperoxaluria has interstitial fibrosis and inflammation that separate crystal-filled tubules. This unique case had marked embryonal hyperplasia characterized by nodular proliferations of small basophilic tubules peppering the cortex; the patient was not dialyzed. Osseous metaplasia is present adjacent to the glomerulus. (H&E; ×250.) |
identified in the responsible gene, CTNS, which resides on chromosome 17p13 and encodes cystinosin, a ubiquitous lysosomal transmembrane protein that facilitates efflux of cystine from the lysosome (203). It differs thereby from other lysosomal storage diseases, which are caused by deficiencies in lysosomal acid hydrolases. Recently identified is a second cystinosin isoform, cystinosin-LKG, that results from alternative splicing and localizes to lysosomes and other cellular compartments including the plasma membrane, endoplasmic reticulum, and small, nonlysosomal cytoplasmic vesicles (204). All mutations currently described alter the sequence of both isoforms; however, the function of cystinosin-LKG remains unknown.
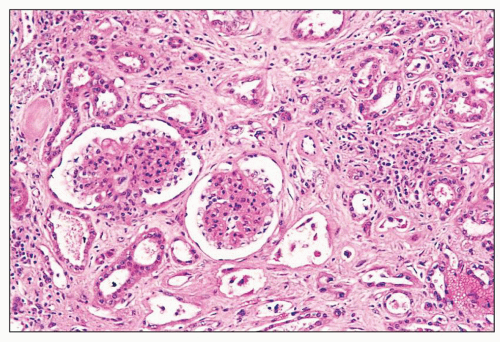 FIGURE 27.34 Renal cortex from an 8-year-old boy with cystinosis showing considerable atrophy of the tubules, interstitial fibrosis, and glomerular solidification. (H&E; ×200.) |
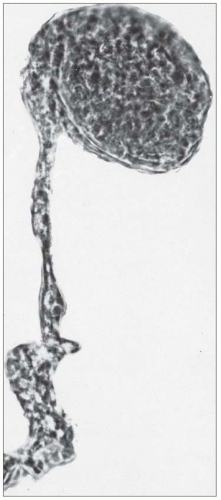 FIGURE 27.35 “Swan-neck” appearance of a dissected proximal convoluted tubule. The thinned early part of the proximal convoluted segment is apparent (glomerulus at top). (Courtesy of Dr. E. M. Darmady.) |
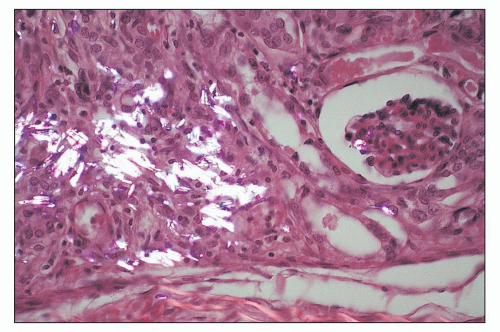 FIGURE 27.36 Alcohol-fixed kidney section from a child with cystinosis showing interstitial deposition of rectangular refractile cystine crystals. A rare crystal is also evident in the glomerulus. (H&E, partial polarization; ×100.) |
are present rarely in tubules and occasionally in the interstitium. The phenomenon is probably caused by a reaction between osmium and intracellular cystine; electron-probe analysis shows that the dark cells contain sulfide, a constituent of cystine. Recently, it was suggested that they represent autophagic vesicles, which are increased in cystinosis (218).
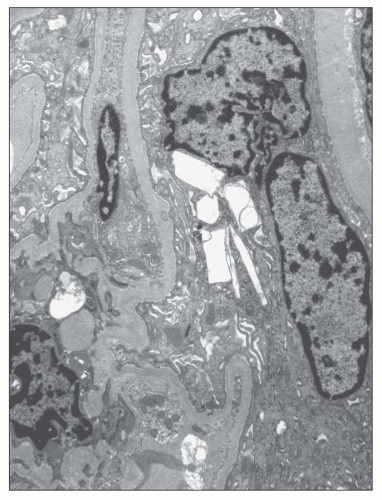 FIGURE 27.37 Electron micrograph shows an epithelial cell from a glomerular tuft that contains rectangular and spindle-shaped clear areas, presumably once occupied by cystine. (×4000.) |
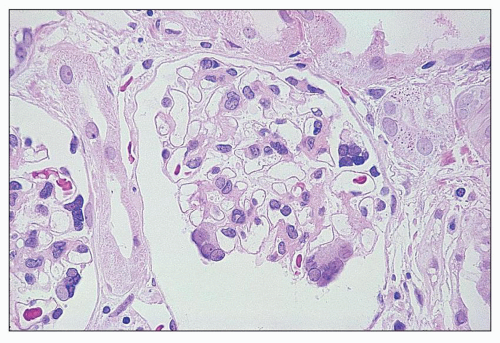 FIGURE 27.38 Glomeruli from a child with cystinosis showing multinucleated visceral epithelial cells. (H&E; ×160.) |
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Glomerular Diseases With Organized Deposits
Glomerular Diseases With Organized Deposits
 Renal Changes With Aging and End-Stage Renal Disease
Renal Changes With Aging and End-Stage Renal Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


