heavy proteinuria and frequent progression to end-stage renal disease (ESRD) (1). Secondary causes of FSGS include genetic defects, viral infections, drugs, toxins, and adaptive responses mediated by altered glomerular hemodynamics (referred to as glomerular hypertension) (Table 6.1). Focal and segmental glomerular scarring also occurs as a nonspecific pattern of injury in the course of progression of diverse inflammatory, proliferative, and thrombotic glomerular diseases.
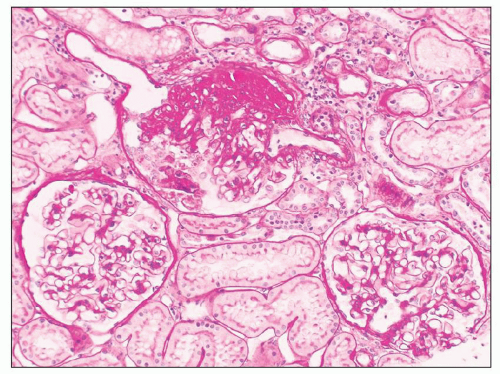 FIGURE 6.1 Primary FSGS. Focal segmental distribution of glomerulosclerosis lesions. The glomerulus at the top shows a discrete segmental scar with adhesion to the Bowman capsule. The two glomeruli at the bottom of the picture are normal. (Periodic acid-Schiff [PAS], ×150.) |
of primary FSGS has grown, and it is now the leading cause of steroid-resistant nephrotic syndrome (SRNS) in both children and adults (10) and the most common primary glomerular disease causing ESRD in the United States (11,12).
TABLE 6.1 Etiologic classification of FSGS | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
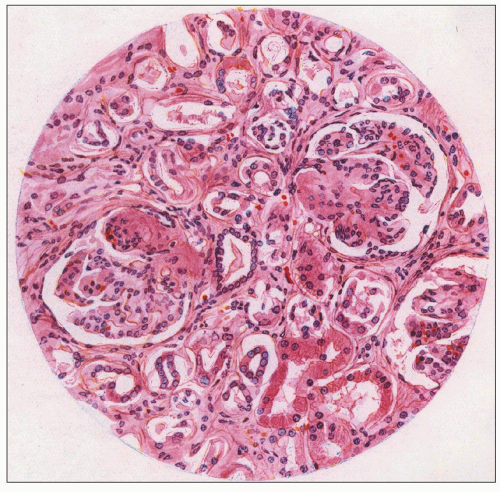 FIGURE 6.2 The first illustration of focal segmental glomerulosclerosis in a patient with nephrotic syndrome (lipoid nephrosis). Two glomeruli show segmental obliteration of capillary lumina with accumulation of matrix. (From Fahr T. Pathologische Anatomie des morbus brightii. In: Henke F, Lubarsch O, eds. Handbuch der speziellen pathologischen Anatomie und Histologie. Berlin: Springer, 1925:156.) |
multiple individual lesions (Fig. 6.8) may exist in a given glomerulus, as revealed by serial sectioning and three-dimensional reconstructions (40). Juxtamedullary glomeruli appear to be more vulnerable to developing FSGS than superficial glomeruli, likely because of their greater single-nephron glomerular filtration rate (GFR) and higher glomerular capillary pressures and flow rates (41,42).
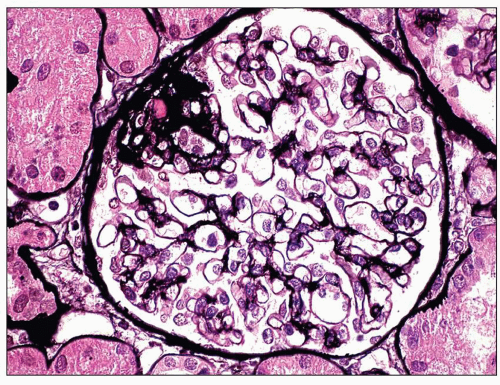 FIGURE 6.3 Primary FSGS, NOS. With a silver stain, the extracellular matrix is argyrophilic (black), whereas hyaline is pink. (Jones methenamine silver [JMS] stain, ×600.) |
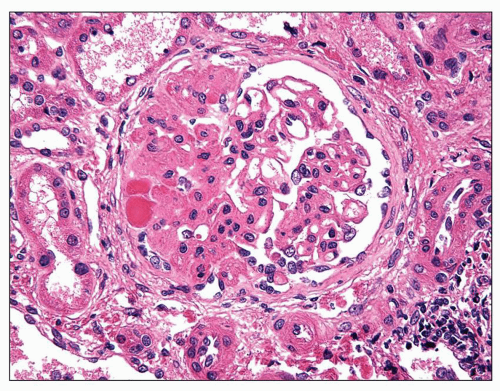 FIGURE 6.4 Primary FSGS, NOS. The left half of the glomerulus shows segmental obliteration of capillary lumina by the matrix and hyalinosis with broad adhesion to the Bowman capsule. Hyaline has a glassy, more eosinophilic appearance than the adjacent matrix material. (H&E, ×400.) |
cells may be seen in areas of segmental sclerosis (Figs. 6.11 and 6.12) and are sometimes numerous, leading to expansion of the glomerular tuft (Fig. 6.13). These cells express monocyte markers, such as CD68, but it is unclear if they derive from circulating macrophages or from transdifferentiation of resident glomerular endothelial or mesangial cells.
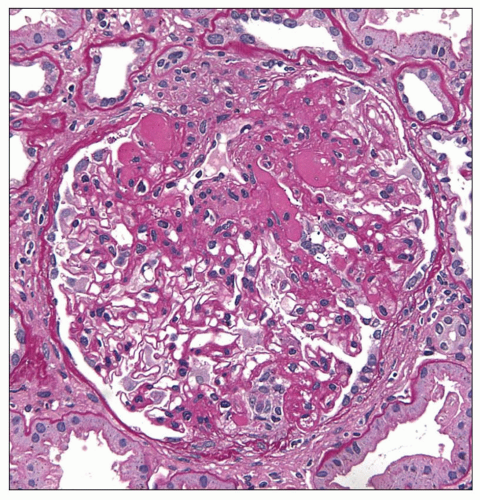 FIGURE 6.5 Primary FSGS. Hyalinosis and matrix both stain pink with PAS, but hyaline has a glassy appearance. (PAS, ×400.) |
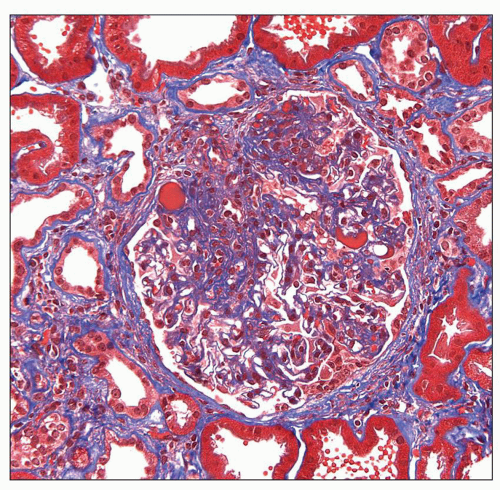 FIGURE 6.6 Primary FSGS. Hyalinosis stains bright red and extracellular matrix stains blue with trichrome stain. (Trichrome stain, ×400.) |
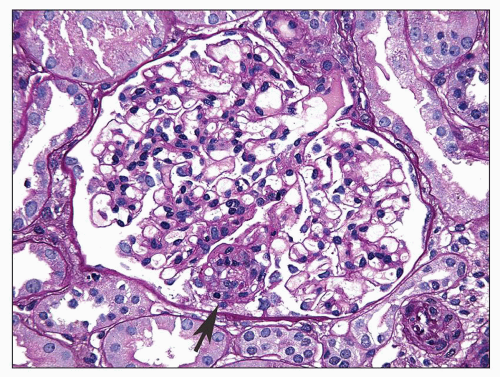 FIGURE 6.7 Primary FSGS. A single segmental lesion (arrow) with endocapillary hypercellularity and mild hyalinosis involves the periphery of the glomerular tuft. (PAS, ×400.) |
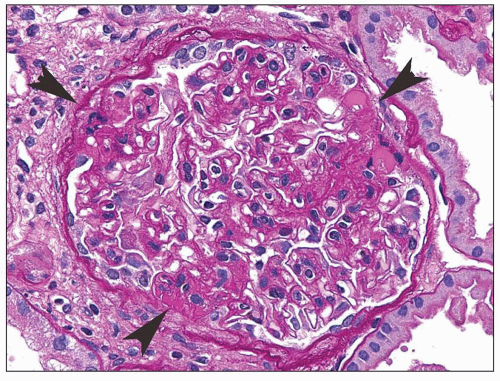 FIGURE 6.8 Primary FSGS. Three discrete lesions (arrowheads) of the matrix and/or hyaline are present in the same glomerulus. (PAS, ×400.) |
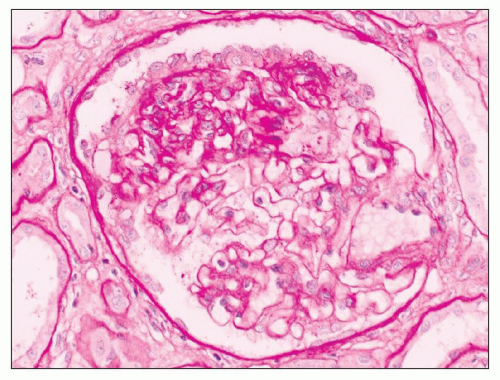 FIGURE 6.9 Primary FSGS. There is a monolayer of podocytes overlying a sclerotic portion of a glomerulus (“cobblestone pattern”). The podocytes are cuboidal with abundant cytoplasm, vesicular nuclei, and prominent nucleoli. (PAS, ×330.) |
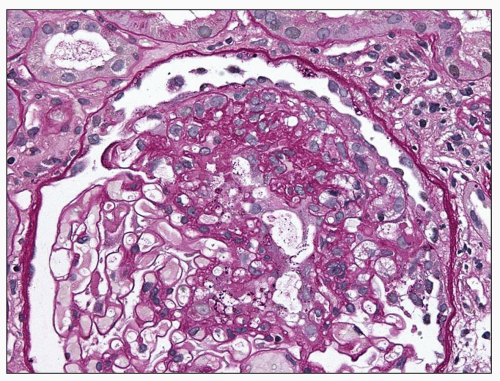 FIGURE 6.10 Primary FSGS. A segmental sclerosis lesion with “capping” of overlying visceral epithelial cells. (PAS, ×400.) |
mild to severe, but are generally commensurate with the degree of glomerulosclerosis (Fig. 6.18). Interstitial foam cells may be seen, either singly or in aggregates, in cases with long-standing proteinuria. In the setting of severe unremitting nephrotic syndrome and marked hypoalbuminemia, proximal tubules may exhibit degenerative and regenerative changes resembling acute tubular necrosis (Fig. 6.19). Arterial vessels show changes related to hypertension and/or aging.
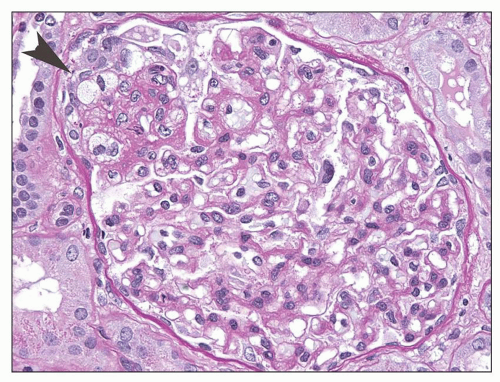 FIGURE 6.11 Primary FSGS. A segmental lesion contains endocapillary foam cells (arrowhead). (PAS, ×400.) |
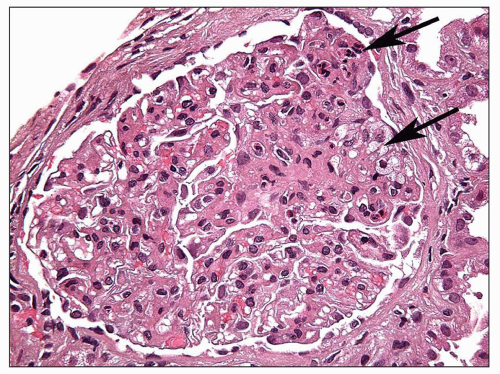 FIGURE 6.12 Primary FSGS. A segmental sclerosis lesion contains endocapillary foam cells, as well as some infiltrating leukocytes (arrows) (H&E, ×400) |
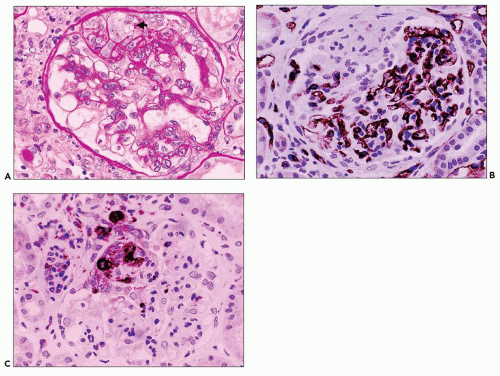 FIGURE 6.13 Primary FSGS with cellular and collapsing features. A: Several glomerular capillaries at the top of the glomerulus are expanded and occluded by foam cells (arrow). The overlying podocytes are hyperplastic. Adjacent capillaries display segmental collapse. (PAS, ×330.) B: The same glomerulus illustrated in (A) is stained for the endothelial marker, CD31. The glomerular capillaries are stained everywhere except in the segmental lesion, indicating that the capillary endothelium has been obliterated. (CD31 immunoperoxidase, ×330.) C: The endocapillary foam cells stain with the macrophage marker, CD68. (CD68 immunoperoxidase, ×330.) |
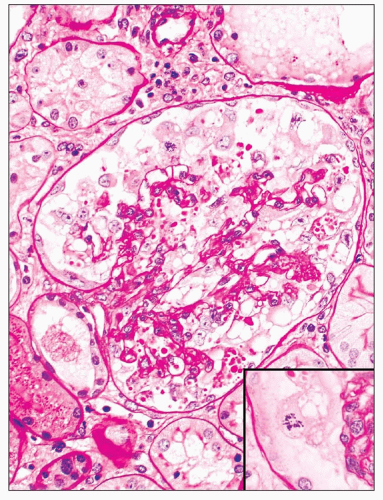 FIGURE 6.14 Primary FSGS, collapsing variant. There is global implosive collapse of capillaries with wrinkling of the glomerular basement membranes, accompanied by swelling and proliferation of glomerular epithelial cells that contain cytoplasmic vacuoles and PAS-positive cytoplasmic droplets. One epithelial cell (inset) displays a mitotic figure. (PAS, ×330.) |
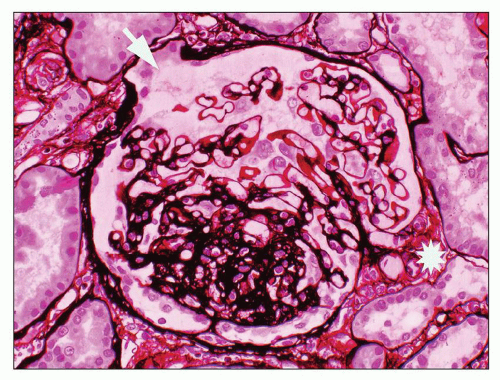 FIGURE 6.15 Primary FSGS, NOS. A segmental sclerosis lesion with a small adhesion involves neither the hilum (asterisk) nor the tubular pole of the glomerulus (arrow). (JMS stain, ×330.) |
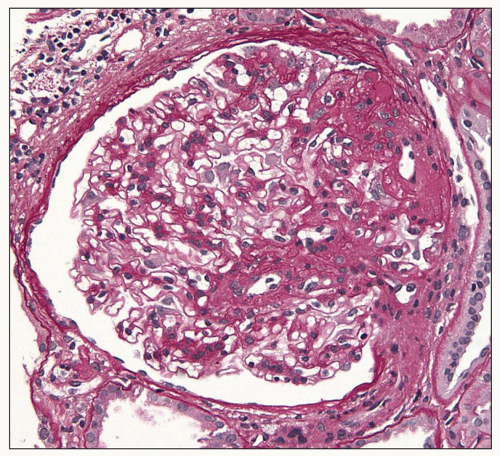 FIGURE 6.16 Primary FSGS, perihilar variant. Segmental sclerosis and hyalinosis located in the perihilar region (vascular pole). (PAS, ×600.) |
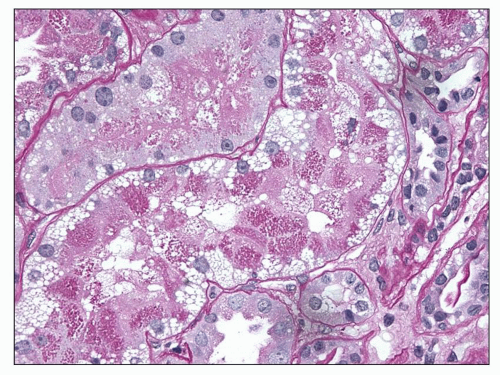 FIGURE 6.17 Proximal tubules display cytoplasmic protein and lipid droplets. (PAS, ×600.) |
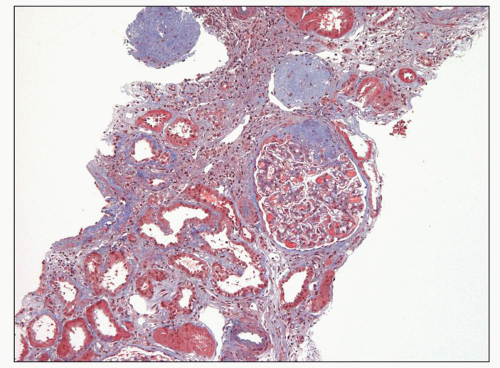 FIGURE 6.18 Primary FSGS. One glomerulus shows segmental sclerosis, and the other two are globally sclerotic, consistent with advanced chronicity. There are prominent tubular atrophy and interstitial fibrosis. (Trichrome stain, ×100.) |
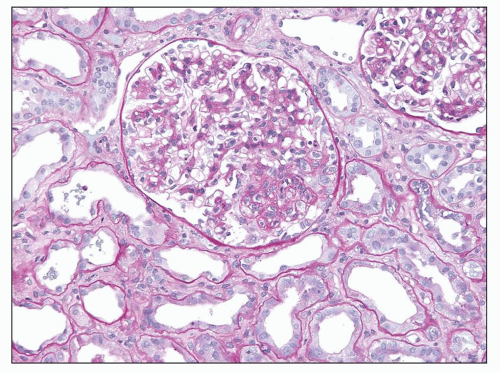 FIGURE 6.19 Primary FSGS, with severe acute tubular injury. Proximal tubules display epithelial simplification with loss of brush border. (PAS, ×200.) |
Many filtration slit diaphragms become displaced or lost. Podocyte actin filaments typically undergo rearrangement to form a dense cytoskeletal “mat” parallel to the direction of the GBM, in the basal cytoplasm above the effaced foot processes. Detachment and lifting of injured podocytes from the GBM may be seen (Fig. 6.23). These changes are often followed by the laying down of a lamellated neomembrane between detached visceral epithelial cells and the underlying GBM. Recent studies suggest that this looser matrix, which is less electron dense than the normal GBM, is derived from parietal cells that have replaced lost podocytes (43). Cytoplasmic electron dense protein droplets are common in cases with severe proteinuria. Hyalinosis lesions are characterized by accumulation of amorphous electron dense material, sometimes containing clear, rounded inclusions representing entrapped lipid and typically localize to the infra-membranous region (i.e., between the endothelial cell and the GBM). Endocapillary foam cells are rounded cells with electron-lucent lipid-rich cytoplasmic vacuolization (Fig. 6.24).
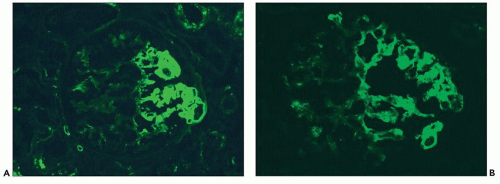 FIGURE 6.20 Immunofluorescence microscopy shows segmental glomerular tuft staining for IgM (A) and C3 (B). (FITC anti-human IgM [A] and FITC anti-human C3 [B], ×330.) |
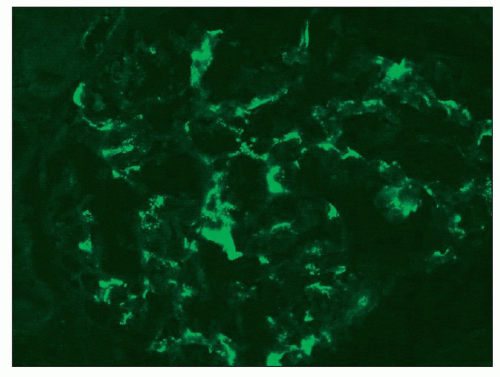 FIGURE 6.21 Immunofluorescence microscopy shows finely granular mesangial staining for IgM. (FITC anti-human IgM, ×330.) |
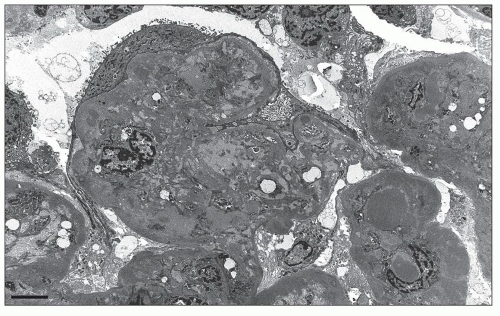 FIGURE 6.22 Primary FSGS. Segmental solidification of the glomerular tuft with wrinkling of basement membranes and inframembranous hyaline is accompanied by diffuse effacement of the overlying podocyte foot processes. (Electron photomicrograph, original magnification ×6000.) |
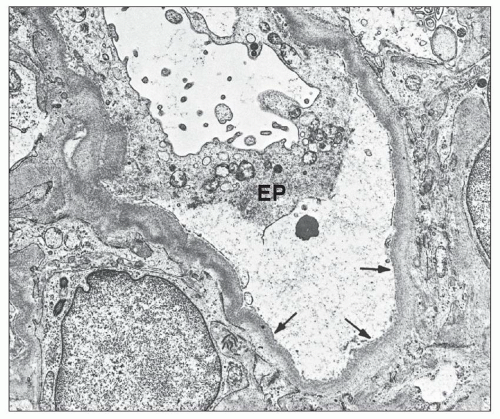 FIGURE 6.23 Electron micrograph shows epithelial cell (EP) detachment from the underlying glomerular capillary basement membrane (arrows). The intervening area contains finely granular material. (Original magnification, ×5000.) (Courtesy of Drs. Jacob Churg and Edith Grishman.) |
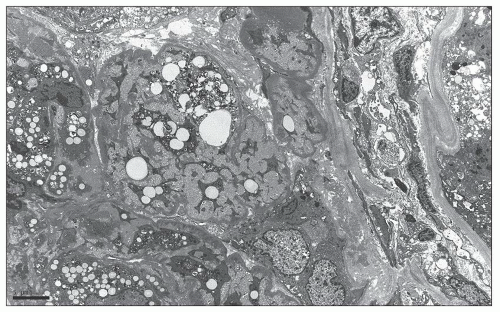 FIGURE 6.24 Primary FSGS, tip variant. Endocapillary foam cells and extracellular matrix obliterate the glomerular capillary lumina. (Electron photomicrograph, original magnification ×3000.) |
some residual patent capillaries,” whereas a global lesion affects the entire glomerular tuft (62). The finding of segmental (Fig. 6.26A) or global (Fig. 6.26B) glomerular capillary collapse with overlying visceral epithelial cell hypertrophy and hyperplasia in at least one glomerulus warrants a diagnosis of the collapsing variant of FSGS, irrespective of the findings in other glomeruli. Excluding collapsing lesions, the finding of a single segmental lesion involving the tip domain (outer 25% of the tuft next to the proximal tubule origin) where the tubular pole is identified is diagnostic of the tip variant (Fig. 6.27). After excluding collapsing and tip lesions, the finding of one glomerulus with segmental expansile endocapillary hypercellularity obliterating capillary lumina (with or without foam cells, hyalinosis, infiltrating leukocytes, karyorrhexis, and epithelial cell hyperplasia) is classified as cellular variant (Figs. 6.13 and 6.28). Perihilar variant is defined as segmental hyalinosis and sclerosis contiguous with the glomerular hilum affecting the majority (≥50%) of glomeruli with segmental lesions, excluding collapsing, cellular, and tip lesions (Fig. 6.29). All other cases are classified as FSGS NOS variant, which by default represents the common, “classic” or generic lesion of segmental sclerosis.
TABLE 6.2 Histologic variants of FSGS | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
matrix accumulation in the acute collapsing lesions, but this may be present in other glomeruli. Hyalinosis lesions are rare in this variant. Capillary collapse is accompanied by prominent epithelial cell hypertrophy and hyperplasia within the Bowman space. The epithelial cells typically line the external surface of the glomerular tuft but may fill the Bowman space, forming a “pseudocrescent” (Fig. 6.31). Serial sections have shown that these lesions are often continuous with the parietal epithelial cell layer; however, these cells usually lack the spindled morphology, intercellular matrix, and pericellular
fibrin seen in true inflammatory crescents. Features of fibrinoid necrosis and ruptures of the GBM or Bowman capsule, which are often identified in crescentic glomerulonephritis, are absent. Visceral epithelial cell nuclei are typically enlarged and show vesicular chromatin and prominent nucleoli; rarely, mitotic figures or binucleated cells are evident (see Fig. 6.25) (56). The swollen epithelial cells often contain large cytoplasmic protein droplets, coarse cytoplasmic vacuoles, and lipid droplets; they may have prominent subpodocyte, tunnel-like spaces that raise the cell bodies off the underlying basement membrane (Fig. 6.32). Proximal tubules frequently show degenerative and regenerative changes (Fig. 6.33), and tubular microcysts (dilated tubules filled with proteinaceous casts) are commonly seen (Fig. 6.34). Tubular atrophy and interstitial fibrosis are often severe and disproportionate to the degree of glomerulosclerosis. Foot process effacement is usually severe. Endothelial tubuloreticular inclusions (TRIs) are not a feature of primary collapsing FSGS but may be seen in secondary collapsing glomerulopathy associated with interferon therapy (71), HIV infection (60), or the podocytopathy of systemic lupus erythematosus (SLE) (72).
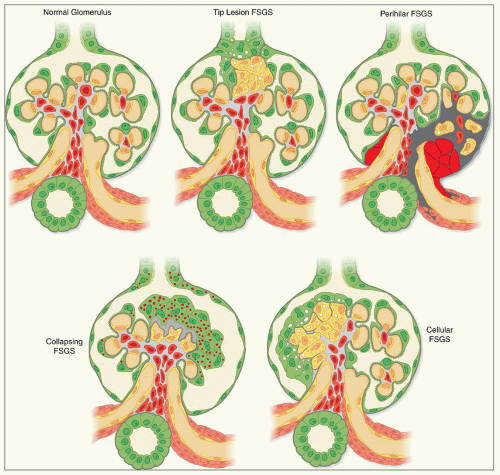 FIGURE 6.25 Drawings of the histologic features of the Columbia Classification variants of FSGS (NOS not shown). Top Row: Normal glomerulus for comparison with epithelial cells green, endothelial cells yellow, mesangial and smooth muscle cells orange, foam cells yellow with clear vacuoles, and basement membranes gray. Tip lesion variant FSGS with segmental lesion involving the tubular pole, confluence of podocytes, and tubular epithelial cells, an endocapillary foam cell (yellow with clear vacuoles) and slight increase in the extracellular matrix. Perihilar variant FSGS with segmental hyalinosis (red) and sclerosis (dark gray) involving the vascular pole (perihilar region). Bottom Row: Collapsing variant FSGS with segmental collapse of capillaries, separation of podocytes from the glomerular basement membrane, accumulation of subepithelial extracellular matrix and hyperplasia and swelling of overlying epithelial cells that have abundant cytoplasmic protein droplets. Cellular variant FSGS with segmental obliteration of capillary lumens by endocapillary hypercellularity caused by foam cells (yellow with vacuoles) and adjacent epithelial hypertrophy and mild hyperplasia. |
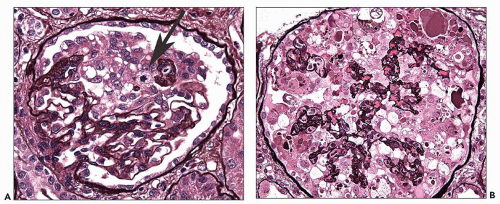 FIGURE 6.26 A: Primary FSGS, collapsing variant. A glomerulus shows segmental collapse of capillaries, associated with marked hyperplasia and swelling of overlying epithelial cells, one of which is undergoing mitosis (arrow). (PAS, ×400.) B: Primary FSGS, collapsing variant. There is global collapse of capillaries, associated with hyperplasia and swelling of overlying epithelial cells, many of which display abundant cytoplasmic protein droplets and vacuoles. (JMS stain, ×400.) |
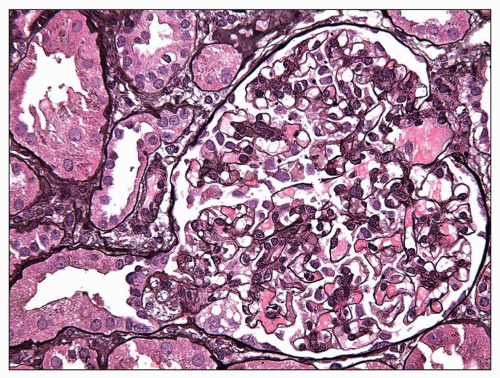 FIGURE 6.27 Primary FSGS, tip variant. Segmental endocapillary foam cell accumulation is seen at the origin of the proximal tubule. The affected segment has prolapsed into the tubular orifice with confluence of overlying podocytes with proximal tubular epithelium. The remainder of the glomerular tuft appears normal in cellularity. (JMS stain, ×400.) |
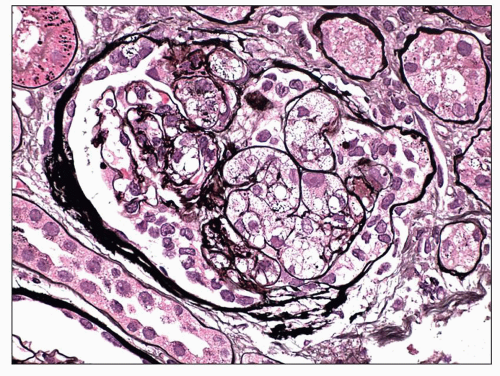 FIGURE 6.28 Primary FSGS, cellular variant. There is segmental expansion of the glomerular tuft by endocapillary foam cells, occluding capillary lumina. (Jones stain, ×600.) |
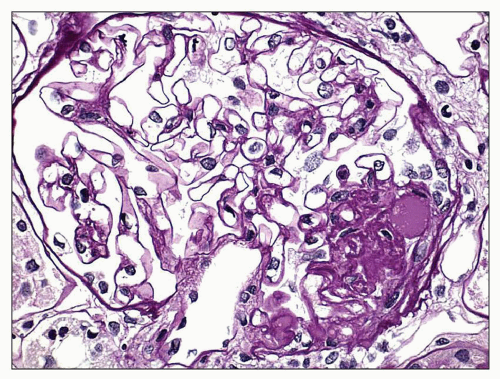 FIGURE 6.29 FSGS, perihilar variant. A glomerulus shows segmental perihilar sclerosis and hyalinosis. (PAS, ×600.) |
tubulointerstitial scarring is not significantly worse in NOS compared to the other variants, implying that NOS is not just an advanced stage of the other variants (64). Thus, the NOS variant may occur ab initio or other variants may evolve into NOS over time.
TABLE 6.3 Frequency of Columbia Classification morphologic variants in primary FSGS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 6.4 Renal outcomes in primary FSGS by histologic variant | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


