traditional classification of MPGN requires modification. A useful classification system should meet the following criteria: (a) define the entity clearly so that the precise connotation of the term can be immediately apparent; (b) be clinically significant, useful, and therapeutically relevant; (c) be based on pathogenesis within the limitation of our current knowledge; and (d) be relatively easy for all to use and morphologically reproducible (119). The traditional pathologic classification of MPGN was based primarily on ultrastructural features and included MPGN type I, MPGN type II, and two variants of MPGN type III. The current classification recognizes the importance of IF (or immunohistochemistry) microscopy in further dividing MPGN into immune complex-mediated MPGN with glomerular immunoglobulins and complement deposition and MPGN with abnormalities in alternative complement pathway regulation resulting in isolated C3 deposits with little or no immunoglobulins by IF. MPGN type II is currently designated DDD and is recognized as a variant of C3 glomerulopathy (see Table 8.1 and Chapter 9).
TABLE 8.1 Pathologic variants of membranoproliferative glomerulonephritis | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
thickening of the glomerular capillary walls, argyrophilic “splitting” of the basement membranes by nonargyrophilic material, and prominent lobulation of the tuft with some central hyaline zones (133,134). Subsequent studies showed similar glomerular lesions in other patients including patients who did not have hypocomplementemia.
TABLE 8.2 MPGN type I associated with known conditions | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
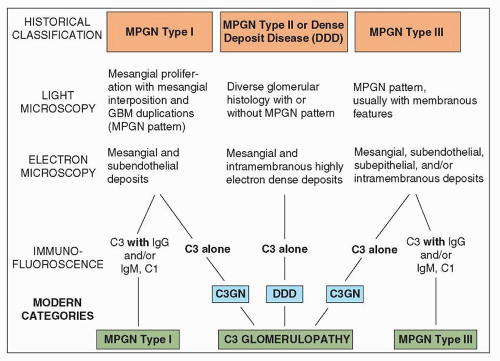 FIGURE 8.1 The evolving classification of membranoproliferative glomerulonephritis. Until recently, the classification of primary MPGN into types I, II, and III was based primarily on histologic features (light microscopy) and the ultrastructural location and electron density of the deposits (electron microscopy). With our increased understanding of the role of complement in the pathogenesis of these conditions, the IF findings now play a crucial role in categorizing MPGN as immunoglobulin-mediated versus non-immunoglobulin-mediated disease; the latter grouping, which is distinguished by isolated C3 staining on IF, has been termed “C3 glomerulopathy.” C3 glomerulopathy encompasses C3 glomerulonephritis (C3GN) and DDD. GBM, glomerular basement membrane; IgG, immunoglobulin G; IgM, immunoglobulin M. (Reproduced from D’Agati VD, Bomback AS. C3 glomerulopathy: what’s in a name? Kidney Int 2012;82:379-381,with permission.) |
hematuria is frequently a finding. MPGN may be asymptomatic and detected only, for example, in school urinary screening of children (152). It has been noted that patients diagnosed with MPGN type I on routine urinary screening usually have lower blood pressure, less proteinuria, and less chronic renal disease when compared to those who are diagnosed when symptomatic, indicating that early identification of the disease by urinary screening may allow for early therapy and improve the prognosis of this disease (152).
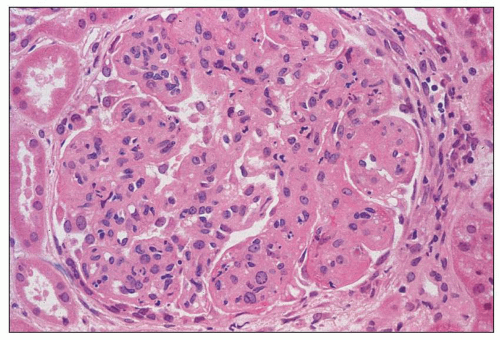 FIGURE 8.2 MPGN type I. There is increased lobulation, intracapillary hypercellularity (including mild neutrophil infiltration), and thickening of the capillary walls. (H&E, ×400.) |
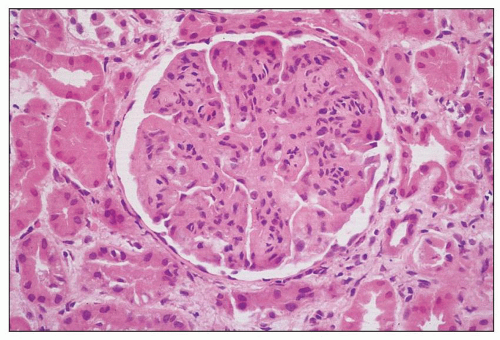 FIGURE 8.3 MPGN type I. There are capillary wall thickenings, increased cellularity, and pronounced lobulation. (H&E, ×360.) |
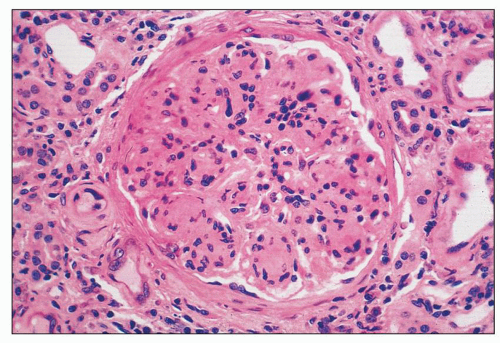 FIGURE 8.4 MPGN type I. Accentuation of the lobular pattern with sclerotic mesangial nodules. (H&E, ×400.) |
severe cases of MPGN. The greatest numbers of monocytes were noted in renal biopsies with the most glomerular hypercellularity and the largest number of glomerular subendothelial and subepithelial deposits. Soma et al. (176) examined the nature of the intraglomerular immune cell infiltration and its relationship to C3 deposits over time. These investigators found monocytes/macrophages and leukocytes to be the predominant cell type at first biopsy (with many fewer T cells). Second biopsy showed either less complement deposition with fewer leukocytes of all types or greater complement deposition with a positive correlation between the number of intraglomerular T cells and monocytes/macrophages (176). Yang et al. (177) and Lan et al. (178), using double immunostaining for CD68 and the proliferating cell nuclear antigen (PCNA), demonstrated that MPGN (presumably type I) is associated with marked macrophage infiltration, with proliferating macrophages (CD68+PCNA+ cells) accounting for up to 42% of total macrophage population. Macrophage proliferation was largely restricted to areas of severe tissue damage (i.e., glomerular hypercellular lesion and foci of tubulointerstitial damage), suggesting that local proliferation is a mechanism for amplifying macrophage-mediated tissue injury. Macrophage accumulation may be partially related to the marked up-regulation of renal expression of macrophage migration inhibitory factor (MIF). In addition, the glomerular and interstitial macrophage proliferation correlated with loss of renal function and histologic lesions but not with proteinuria. In a recent study, Wu et al. (179) characterized and quantified the proliferating cells in MPGN (presumably type I) using monoclonal antibodies for various cell markers. They demonstrated marked mesangial proliferation/activation coupled with increased neutrophils, macrophages, and T cells. However, endothelial cell proliferation was not obvious. Cases of primary mixed or essential cryoglobulinemic glomerulonephritis with a membranoproliferative glomerular pattern generally have a large number of infiltrating monocytes and macrophages (180,181).
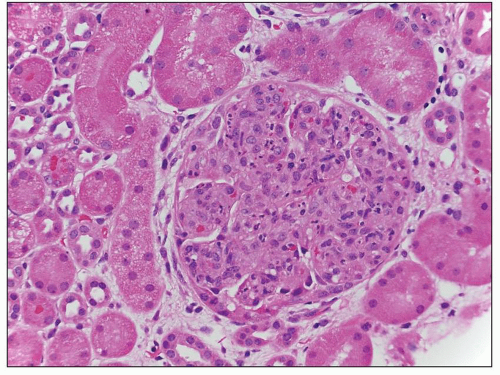 FIGURE 8.5 MPGN type I. There are numerous infiltrating neutrophils (exudative form) resembling acute postinfectious glomerulonephritis by light microscopy. (H&E, ×400.) |
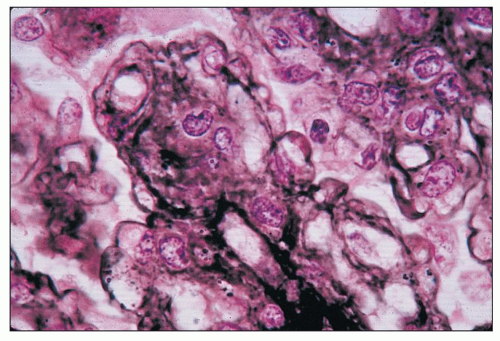 FIGURE 8.6 MPGN type I. Glomerulus with silver stain shows tram tracking or reduplication of the GBM. (Jones silver methenamine, ×600.) |
scattered glomerular fuchsinophilic subepithelial humps may be detected with trichrome stains and oil immersion. Bohle et al. (186) have suggested that “hyperperfusion injury” can be seen with great frequency in MPGN. This is defined as the presence of glomerular adhesions (synechiae), glomerular subendothelial hyalinosis, and fat droplets in the hyaline material.
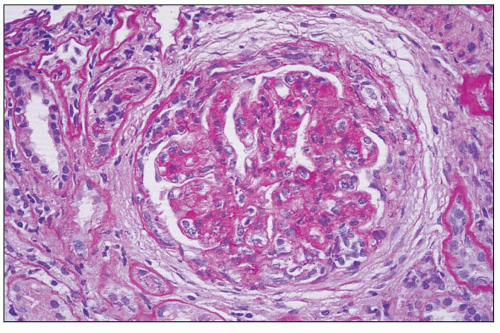 FIGURE 8.7 MPGN type I. Glomerulus from a case of MPGN type I with crescent. (PAS, ×400.) |
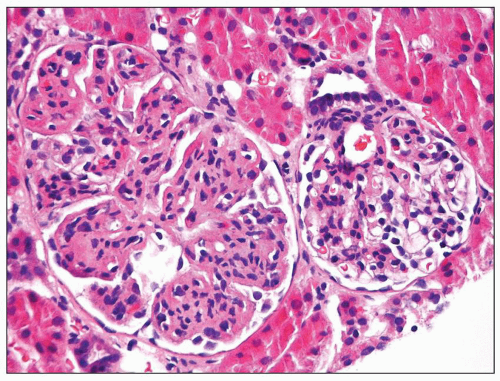 FIGURE 8.8 Focal MPGN type I. One glomerulus shows typical changes of MPGN type I, while the other glomerulus reveals only mild mesangial hypercellularity. (H&E, ×400.) |
immunoglobulin deposition (52). Conspicuous IgM or IgA even in the absence of IgG is indicative of an immune complex MPGN rather than a C3 glomerulopathy variant of MPGN.
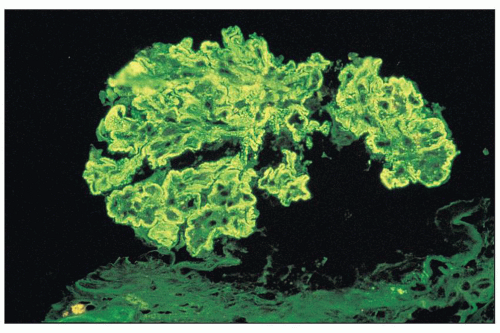 FIGURE 8.9 Immunofluorescence of MPGN type I. There is intense glomerular mesangial and capillary wall staining with anti-IgG antiserum. (×400.) |
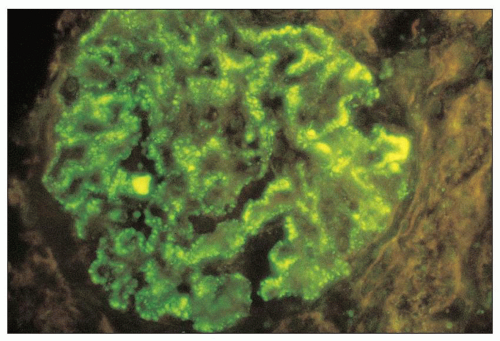 FIGURE 8.10 Immunofluorescence of MPGN type I. There is moderately intense staining for IgM along the glomerular capillary walls in a granular pattern. (×400.) (Courtesy of Dr. Zoltan Laszik.) |
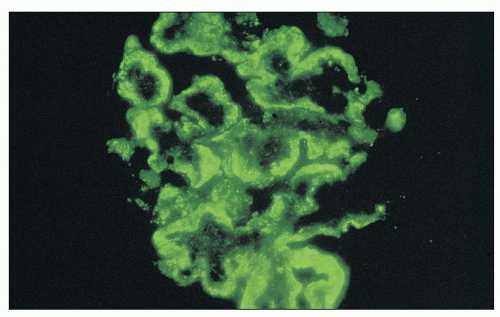 FIGURE 8.11 Immunofluorescence of MPGN type I. There is intense staining aong all glomerular capillary walls for C3. The granular staining is quite broad in this instance. (×400.) |
range from small and discrete to large and elongated; they are found both along the periphery of the filtering glomerular capillary walls and just under the paramesangial GBM as it overlies the mesangium. Discrete electron-dense deposits also may be found within the mesangium; at this site, they are usually small, but sometimes, they are more bulky. They are associated with an increase in mesangial or endocapillary hypercellularity (see Fig. 8.16). Sometimes, the IF discloses intense and widespread positive staining along the capillary walls, whereas only scant deposits are present on the EM.
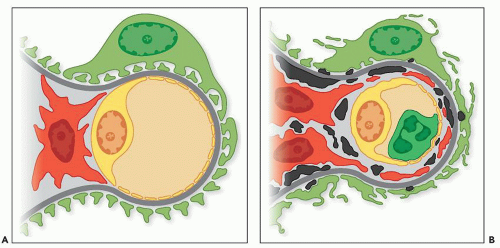 FIGURE 8.12 Drawing depicting a normal glomerular capillary and adjacent mesangium (A) compared to the ultrastructural changes of MPGN type I (B). Note the subendothelial, mesangial and few subepithelial dense deposits (black), capillary wall mesangial interposition (red), new layers of subendothelial matrix material (gray), and increase in mesangial cell numbers. |
a few patients for whom there has been long-term follow-up (124,140,148). The series of Levy et al. (140) showed complete remission in only 4 of 84 children. In this study, 17 children went into remission, but 13 relapsed; in only 4 patients was the remission maintained for periods of up to 4 years (140). Kim et al. (139) noted complete clinical remission in 5 of 63 patients.
Related posts:
 Development of the Kidney
Development of the Kidney
 Focal Segmental Glomerulosclerosis
Focal Segmental Glomerulosclerosis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


