both classifications use diagnostic criteria of renal impairment (urine output, rise in sCr) that manifest at a late stage of injury and rely on knowledge of baseline creatinine (7). Inclusion of other more sensitive criteria, including biomarkers, may enhance definition criteria (8). There has also been lack of precision in the use of the term acute tubular injury/necrosis. The term should be reserved for the clinical pathologic entity of intrinsic renal failure that is the result of either an ischemic or toxic insult to the kidney, with evidence of tubular injury/dysfunction such as altered fractional excretion of sodium (8) and potentially other more specific biomarkers, when other causes have been excluded. As a result of the lack of uniformity in terminology, the percentage of cases of ARF that can be attributed to “acute tubular necrosis/injury” are difficult to accurately ascertain, but the condition is likely responsible for the majority of cases of ARF that require acute renal replacement therapy. The term acute tubular necrosis itself is a misnomer, since necrosis, while classically a feature of animal models, is only one morphologic manifestation of clinical ATI. It should also be noted that morphologic evidence of frank tubular necrosis is not a frequent feature in kidney biopsies obtained in the context of clinical ARF; however, morphologic changes of sublethal tubular injury are usually present. Just as in the clinical classifications, however, morphologic signs of injury appear in a later stage of injury; more sensitive markers are required to identify early tubular cell injury. The term acute tubular injury is more accurate and will be used throughout this chapter.
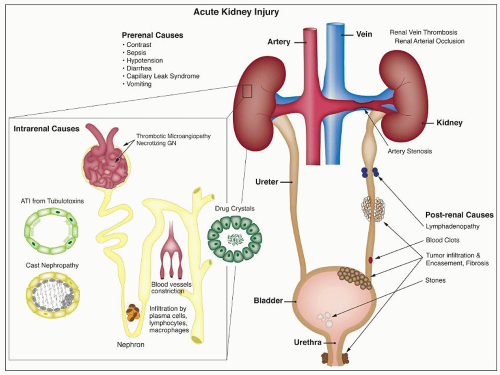 FIGURE 26.1 Causes of acute renal failure. This figure depicts several etiologies that may lead to AKI. Intrarenal causes of tubular injury include tubulotoxins, light chain casts, ischemic injury, and lesions that indirectly affect tubular cell viability through impaired glomerular blood flow (i.e., TMA and necrotizing GN). Other common causes of ATI include drug crystals with associated tubular obstruction, interstitial inflammatory infiltrate by lymphocytes, plasma cells, and macrophages. Prerenal causes include hypotension, diarrhea, vomiting, and contrast. Postrenal causes include lymphadenopathy, tumor infiltration, and urinary outflow obstruction by fibrosis and blood clots and stones. |
thereafter, Baker and Dodds (11) studied a rabbit model of ARF and emphasized tubular obstruction as important in the pathogenesis. Progress in the field was relatively dormant until the advent of World War II, when Bywaters and Beall (12) revived interest in ARF as a result of their study of London air casualties. Bywaters and Dible (13) and Dunn et al. (14) described intratubular hemoglobin and myoglobin casts associated with focal necrosis of tubules, interstitial edema, and mild interstitial inflammation localized to specific portions of the nephron in the “crush syndrome or traumatic anuria.” This finding led to the hypothesis that tubular obstruction by necrotic debris and precipitated pigment was the prime cause of the observed oliguria.
TABLE 26.1 Clinical phases of AKI | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
flow distribution demonstrated that a diminution of outer cortical flow correlated best with decreased glomerular filtration (29,30). These and other studies led to a proposal that tubular epithelial injury induced either by ischemia or by a toxin could be sublethal but had to be severe enough to result in decreased epithelial transport activity, which then would result in decreased tubular sodium reabsorption and local activation of the renin-angiotensin system. This, in turn, would alter glomerular hemodynamics and result in decreased glomerular filtration. Decreased tubular urine flow associated with the shedding of cellular debris and the presence of Tamm-Horsfall protein would result in tubular obstruction. When combined with focal areas of necrosis, as demonstrated by the microdissection studies of Oliver, this could lead to a back leakage of fluid, all of which contribute to the end result of oliguria. The term acute renal success was suggested by Thurau and Boylan (31), interpreting the pathophysiologic changes of decreased glomerular filtration as a defense against loss of intravascular volume caused by the inability of the damaged tubules to reabsorb the glomerular filtrate.
studies, but only a minority progress to overt renal failure. A few tubular toxins cause injury only at high doses; with other agents, some level of injury can be detected at the usual therapeutic doses in most patients. Pigmented casts or crystals may appear in the urine, providing a clue to the diagnosis; in such cases, oliguria and even anuria may be the presenting feature. Hydration and maintenance of diuresis help prevent renal dysfunction or hasten recovery in cases with intratubular crystals or cast formation. Radiographic studies generally reveal normal-sized kidneys with increased echogenicity. Clinical features of major nephrotoxins are described in the text following; the focus of this chapter is on toxic effects of therapeutic agents. Toxic nephropathies caused by heavy metals and other environmental and food toxins have been reviewed (40).
TABLE 26.2 Drugs that are injurious to renal tubular epithelium | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
with monitoring of trough levels may enable avoidance of significant renal toxicity, even in elderly patients (64). However, with prolonged treatment, differences between once-daily and twice-daily dosing diminish (65). The toxicity of aminoglycosides may be potentiated by ischemia or other drugs, including thalidomide (66).
defined to include an asymptomatic mild decline in the GFR, it is likely that many patients treated with immunosuppressive doses of CsA experience nephrotoxicity. When more overt CsA-induced renal failure is superimposed on mild functional toxicity, it may manifest in the form of one or more clinical syndromes: acute reversible renal functional impairment, delayed renal allograft function, tubular cell effects, acute vasculopathy (thrombotic microangiopathy), and chronic nephropathy with interstitial fibrosis.
contrast dose, 5-mL contrast volume × body wgt (kg)/baseline sCr (mg/dL), has been developed (157). Several recent studies confirm that exceeding this threshold increases the risk for contrast-induced nephropathy (158,159), at least in percutaneous coronary intervention studies. Patients who develop RN reportedly have more frequent adverse events, including myocardial infarction, prolonged hospital stay, worse kidney function at discharge, and higher mortality (153,160).
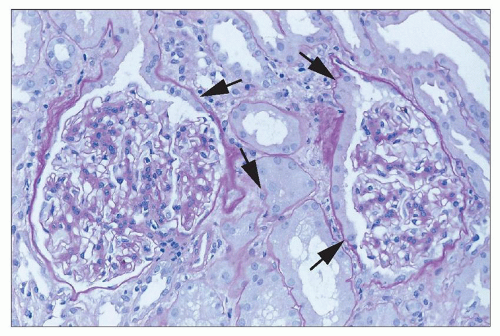 FIGURE 26.2 “Tubularization” of parietal epithelial cells lining the Bowman capsule (arrows). Reactive changes in the proximal tubule extend from the tubular takeoff to involve these epithelial cells, which have marked increase in cytoplasm compared to normal quiescent cells. (H&E; ×640.) |
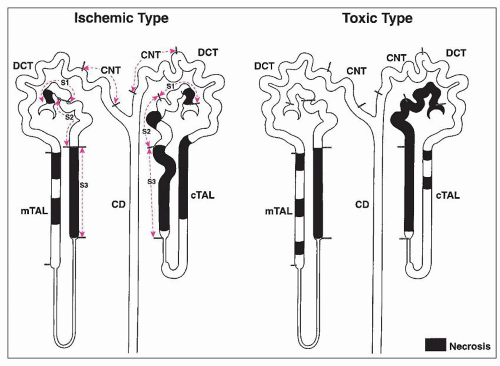 FIGURE 26.3 Cartoon demonstrating the difference in the distribution of lesions between ischemic and classic nephrotoxic tubular injury. In addition to differences in localization along nephron segments, different degrees of damage are visible between cortical and juxtamedullary nephrons. In the ischemic form, the S3 segments are most severely affected, along with focal areas of the ascending limbs of the loop of Henle. The cortical nephrons show more extensive damage than the juxtamedullary nephrons. In the toxic form, tubular epithelial damage is more extensive. Whereas mercury shows some predilection for the S3 segment, other heavy metals and organic toxins often show more extensive involvement of all nephron segments, also with a greater predilection for cortical nephrons. |
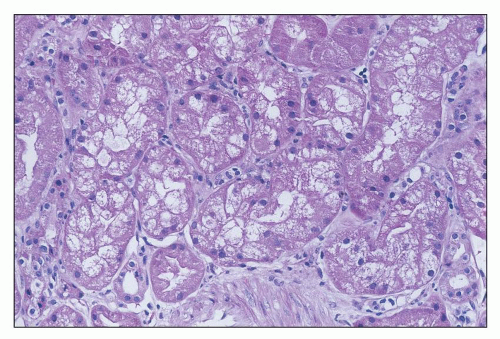 FIGURE 26.4 Tubular cells showing severe cell swelling, in some areas apparently obstructing the tubular lumen. (H&E; ×640.) |
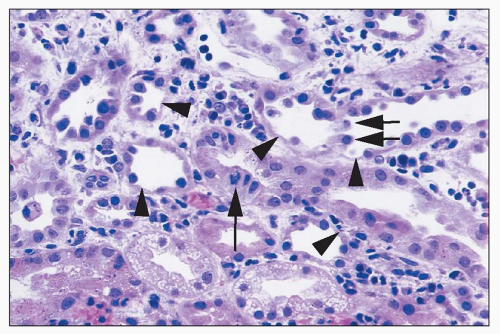 FIGURE 26.5 Areas of single tubular cell loss from a kidney with ischemic injury. Injured cells have detached, leaving areas of tubular basement membrane covered by a thin layer of cytoplasm from adjacent cells (arrowheads). A few detached cells can be seen in tubular lumina (short arrows). A mitotic figure can be seen in one tubular cell (long arrow). There are also interstitial edema and inflammatory cells largely marginating in capillaries. (H&E; ×400.) |
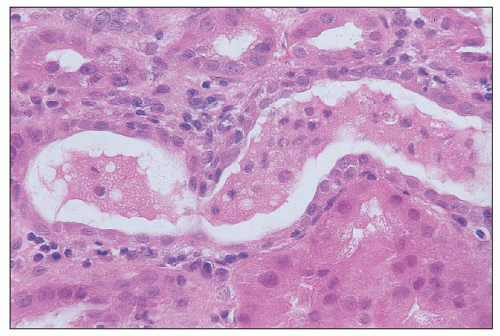 FIGURE 26.6 Intact exfoliated tubular cells in tubular lumen in a kidney with ischemic injury. (H&E; ×640.) |
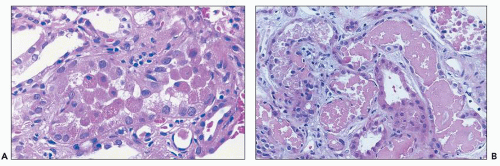 FIGURE 26.7 A: Detached necrotic tubular cells, several with pyknotic nuclei, in the lumen of proximal tubule. B: Granular casts with necrotic cell debris. Note flattened tubular epithelium in tubules containing necrotic debris. (H&E, ×640.) |
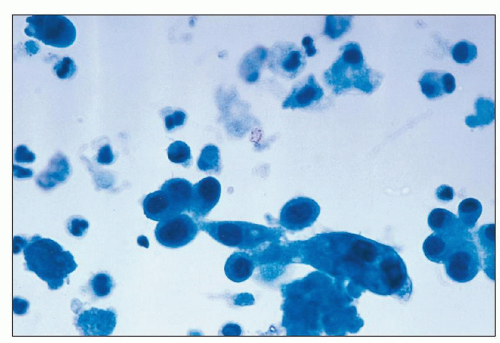 FIGURE 26.8 Intact tubular cells in the urine from a patient with ischemic injury. (Papanicolaou; ×1,000.) |
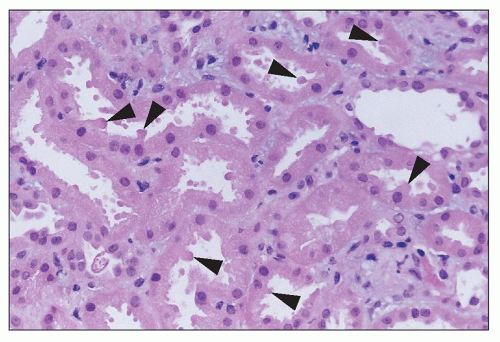 FIGURE 26.9 Apical blebbing from the surface of injured proximal tubular cells (arrowheads). Apical cytoplasmic blebs can be seen in tubular lumina. (H&E; ×640.) |
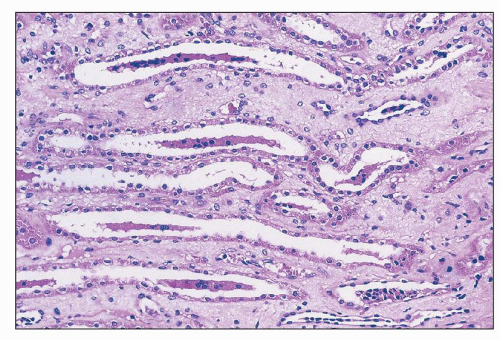 FIGURE 26.10 Tubular cell casts in collecting ducts in papilla. Injured cells have detached from sites in proximal nephron and aggregate into casts, often around a protein core. (H&E; ×400.) |
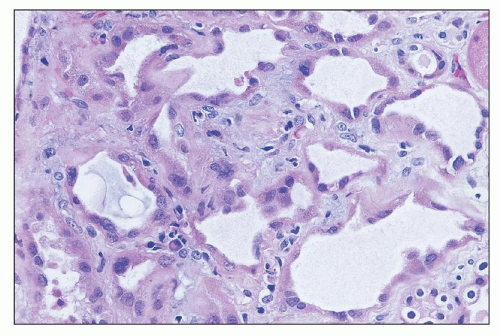 FIGURE 26.11 Dilated tubules with flattened epithelium in the regenerative phase after tubular injury. Marginating inflammatory cells can be seen in capillaries. (H&E; ×640.) |
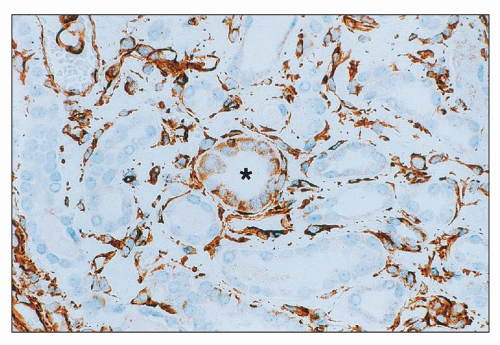 FIGURE 26.12 Injured tubule with cells staining for vimentin (center). Adjacent tubules do not stain. Note bright background staining for vimentin in interstitial areas. (Immunoperoxidase; ×400.) |
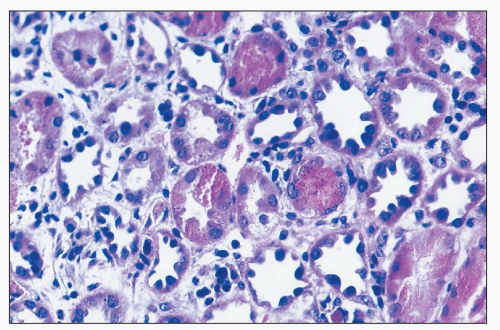 FIGURE 26.13 Striking regenerative changes in tubular cells, with pleomorphic hyperchromatic nuclei, with relatively little cytoplasm. (H&E; ×640.) |
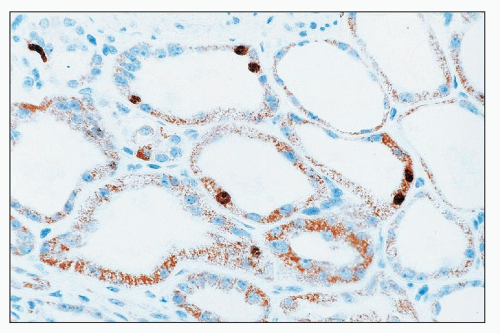 FIGURE 26.14 Immunostain for the transcription factor Ki-67. Note positive nuclear staining in several tubular cells. (Immunoperoxidase; ×640.) |
with nuclear fragmentation; and a variety of other cytopathic changes leading to necrosis (Fig. 26.18). Detachment of tubular epithelial cells may be seen (Fig. 26.19).
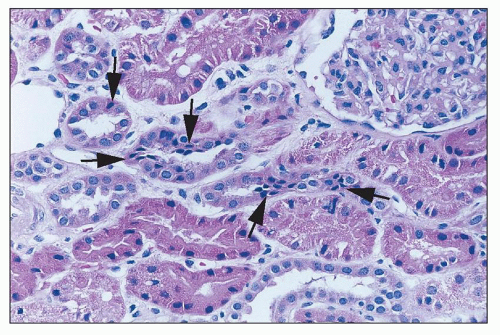 FIGURE 26.15 Focal tubular cell apoptosis, with condensed triangular cells in the epithelium (arrows), focally extruding from the epithelium. (H&E; ×640.) |
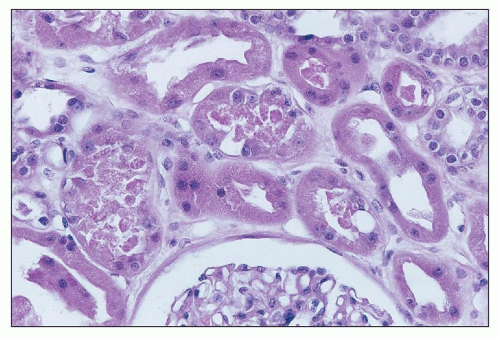 FIGURE 26.16 Coagulative necrosis of focal tubular epithelial cells, with cell debris in the tubular lumina. (H&E; ×640.) |
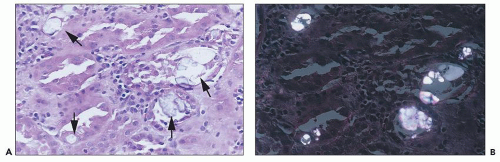 FIGURE 26.17 A: Oxalate crystal precipitates (arrows) in ATI in an allograft kidney. B: Oxalate crystals in polarized light. (H&E; ×640.) |
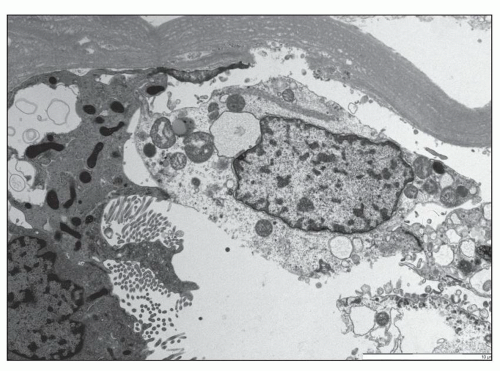 FIGURE 26.18 An electron micrograph shows a tubular epithelial cell in the process of desquamation into the lumen. The cell has separated from the basement membrane but is still adherent to the adjacent epithelial cell through the cell-to-cell junctions. (×3,000.) |
has been proposed as a sensitive early marker of AKI, with markedly increased expression (199,200). The L-1 cell adhesion molecule has been identified as a potential biomarker of distal tubular injury in AKI, with loss of normal polarized distribution in the collecting duct, and induction of expression in medullary thick ascending limb and distal tubule with injury (201). Apoptotic cell death, difficult to detect by histology, can be detected in tissue using techniques such as terminal deoxynucleotidyl transferase dUPT nick end labeling (TUNEL) (201). An increase in progenitor cells (e.g., CD133+ CD24+ CD106- cells) can be identified in tissue sections by immunostaining as a correlate of injury (202). Other potential injury markers in tissues are described below in the section on “Pathophysiology.”
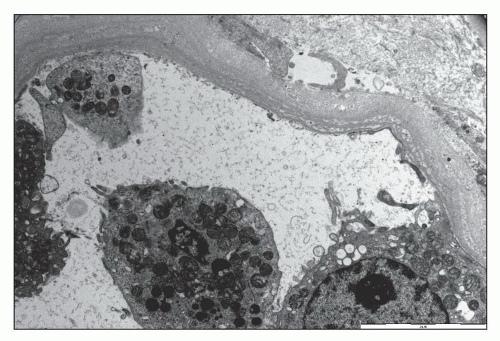 FIGURE 26.19 An electron micrograph demonstrates an area of loss of an array of tubular epithelial cells. (×3,000.) |
Alterations in the surface of the cells, including loss of brush border (detectable on PAS), loss of basolateral infoldings, and blebbing of apical cytoplasm
Cytoplasmic swelling and vacuolation
Intracellular inclusions
Extensive tubular cell necrosis
Loss of individual tubular cells, with gaps along the tubular basement membrane or tubular profiles with fewer and attenuated cells lining the tubule
Intraluminal proteinaceous cellular debris, casts, or crystals
Tubular dilation with flattening of tubular epithelium
Tubular rupture with urinary extravasation
Regenerative changes, including flattening of epithelial cells, cytoplasmic basophilia, heterogeneity in cell size and shape, a higher nuclear-to-cytoplasmic ratio in individual cells, and cellular mitoses
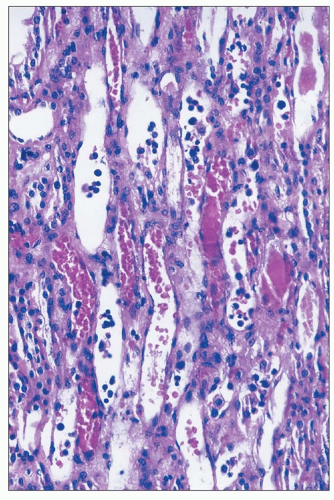 FIGURE 26.20 Erythrocyte congestion and nucleated cells in dilated vasa recta in the outer medulla of a kidney with ischemic injury. (H&E; ×400.) |
injury (207). It is clear that both necrosis and apoptosis occur in toxic nephropathies in experimental models as well as in clinical tubular injury (208,209). In vitro studies have documented apoptosis in cell culture on exposure to nephrotoxins. For example, LLC-PK1 cells exposed to sublethal doses of mercuric chloride in vitro undergo apoptosis (210), and apoptosis can be induced in Lewis lung cancer-porcine kidney-1 [LLC-PK1] cells by cisplatin as well via activation of caspases (211). Our understanding of the role of apoptosis in the pathogenesis of toxic renal injury has evolved over the past decade. ATI due to cisplatin therapy depends also partially on Fas-mediated apoptosis driven by Fas ligand (FasL) expressed on tubular epithelial cells (212). Moreover, cisplatin down-regulates the expression of the taurine transporter gene TauT in LLC-PK1 cells (213). Taurine is one of the organic osmolytes, which have important antiapoptotic properties in the kidney (214). The antiapoptotic function of organic osmolytes in kidney cells is mediated through suppression of efflux of proapoptotic molecules, such as cytochrome c, from the mitochondria.
especially with the nucleotide reverse transcriptase inhibitors adefovir and tenofovir, with degenerative changes, thinning and vacuolization of cytoplasm, loss of brush border, and even tubular cell necrosis, with nuclear changes reminiscent of viral inclusions (61,219,220,221). On electron microscopy, alterations in mitochondria have been observed, with swelling and dysmorphic changes. Changes include variable mitochondrial size, with some small and rounded and others swollen with irregular contours, clumping, loss and disorientation of cristae, and focal marked reduction of mitochondria (61). Giant mitochondria, in some cases, the size of nuclei, fuchsinophilic on trichrome stain but PAS- and silver-negative, may be seen in proximal tubular epithelial cells. Patchy interstitial inflammation has been described without crystals (222), and, occasionally, granulomas have been described. Renal tubular cell apoptosis has been detected in renal biopsy of a patient with irreversible cidofovir toxicity (223), and fibrosis and tubular atrophy have been described with tenofovir (61), with persistent renal dysfunction.
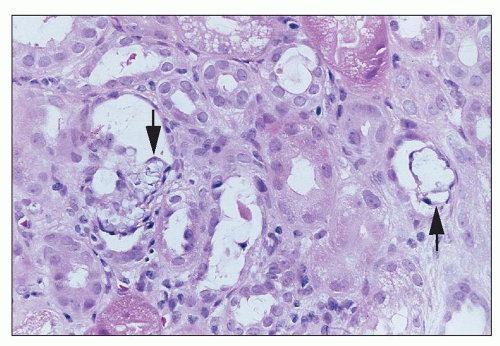 FIGURE 26.21 Crystalline precipitates (arrows) in tubules in a patient treated with intravenous acyclovir. (H&E; ×640.) |
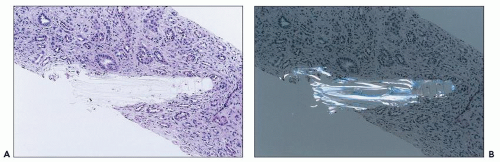 FIGURE 26.22 A: Indinavir crystal in papilla of a patient treated long term with highly active antiretroviral therapy. The patient also had crystals in the urine. B: Indinavir crystal under polarized light. (H&E; ×200.) |
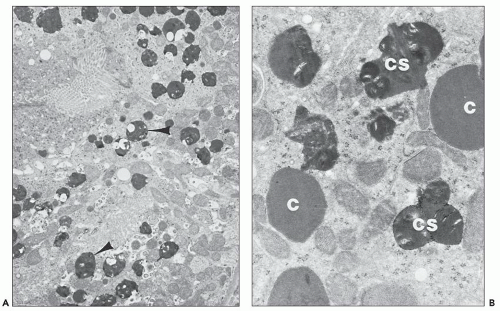 FIGURE 26.23 Electron micrographs of a rabbit killed 14 days after being given a high dose of gentamicin. A: Cytosegresomes (arrowheads) are scattered throughout the cytoplasm of the proximal convoluted tubule. The brush border can be seen at the top left (×5,700). B: Numerous cytosomes (C) and cytosegresomes (CS) are visible at higher power (×2,800) (lead citrate and uranyl acetate). (Courtesy of Dr. E. F. Cuppage.) |
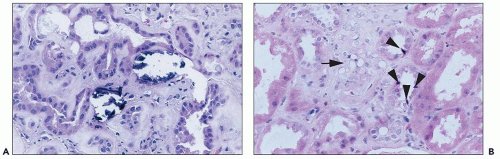 FIGURE 26.24 A: Calcification of tubular cells in a patient treated with amphotericin. B: Striking vacuolization of smooth muscle cells (arrow) in small vessels in the biopsy of a patient treated with amphotericin. Note apoptotic cells (arrowheads) in adjacent tubules. (H&E; ×640.) |
child with elevated vancomycin levels and ARF revealed focal tubular dilation with attenuation of brush border, hyaline casts, and one neutrophil cast without interstitial nephritis (239).
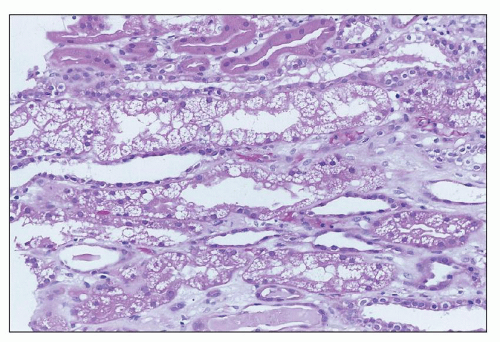 FIGURE 26.25 “Isometric” vacuolization, with many small equal-sized vacuoles in tubular cell cytoplasm, in a patient with high serum levels of calcineurin inhibitor. (H&E; ×400.) |
medullary rays and the inner stripe of the outer medulla. Tacrolimus alone produced increased juxtaglomerular apparatus granularity.
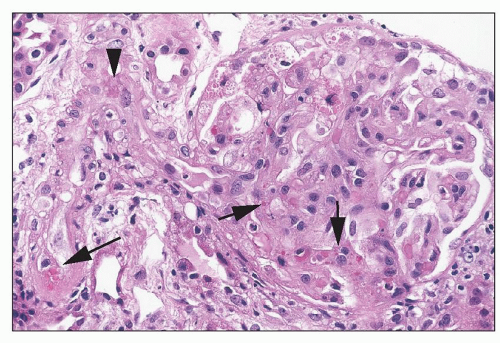 FIGURE 26.26 Thrombotic microangiopathy in a patient on tacrolimus. Note the arteriole with very focal intramural fibrin (arrowhead), focal erythrocyte extravasation into the intima (long arrow), and focal erythrocyte fragmentation in the glomerulus (short arrows). (H&E; ×640.) |
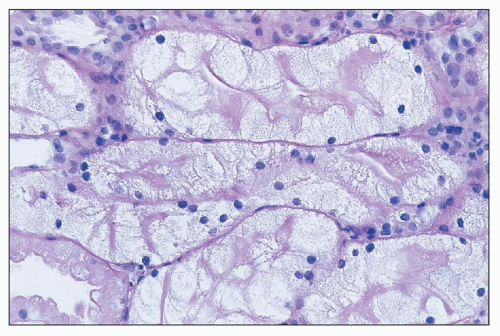 FIGURE 26.27 Severe cell swelling of tubular cells in the biopsy of a patient being treated with IVIG. Note persistence of brush border. (H&E; ×640.) |
ATN has been reported only rarely in patients (265,267,270), recent studies have shown that radiocontrast agents induce apoptosis in proximal tubule cells (271). Increased ceramide synthesis, which stimulates apoptosis, is an important contributing factor to radiocontrast-mediated nephropathy (272).
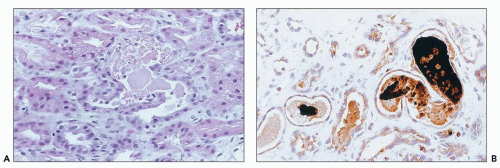 FIGURE 26.28 Pigmented casts in the tubular lumina of a biopsy from a patient with ARF who had overdosed on cocaine. A: Light microscopy. (H&E; ×640). B: Immunostain for myoglobin. (Immunoperoxidase; ×640.) |
In the postnatal period, hypoxic/ischemic injury and toxins are the most common etiologies. Toxic ARF is most commonly associated with administration of aminoglycoside antibiotics and NSAIDs given to close a patent ductus arteriosus. Decreased renal function can be documented in about 40% of premature infants receiving indomethacin; the decrease is usually reversible.
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Glomerular Diseases With Organized Deposits
Glomerular Diseases With Organized Deposits
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


