CYSTIC KIDNEY DISEASES
Cystic kidney diseases are heterogeneous in origin, distribution, and pathogenesis. Cysts usually originate in the tubules but may also involve the glomeruli. Depending on patient age and or heredity, cysts are focal or diffuse, unilateral or bilateral, occur as an isolated defect, or accompanied by cysts in other organs. For example, cysts in the liver are common in autosomal dominant polycystic kidney disease (ADPKD).
Most kidney cysts are benign; some are malignant or neoplastic with malignant potential. Genetic studies reveal an increasing number of mutations accompanying kidney cysts.
Furthermore, mutations are now detected in entities previously thought to be sporadic (e.g., multicystic renal dysplasia and multilocular cyst/cystic nephroma). Early pathologic classifications were based mainly on morphologic characteristics (
1) or incorporated clinical, radiologic, and genetic criteria (
2). More recently, some authors proposed to classify cystic kidney diseases based exclusively on pathogenesis, for example, under “ciliopathies.” This option follows new discoveries that show protein products of gene mutation causing cysts to be localized on cilia or their basal bodies. There are two types of cilia: motile and nonmotile or primary cilia, which are ubiquitous on eukaryotic cells, including renal tubular epithelial cells (
3). Which classification will prevail in the future remains to be seen, but the success of a classification scheme is generally measured by its precision (the degree of observer variability in assigning a specific case to a specific category) and accuracy (the degree to which a specific category correlates with specific defining features). The cilia classification is conceptually more precise; however, not all cysts have a genetic cause; therefore, accurate grouping of the latter entities is not possible on the basis of genetics alone (
4). In addition, the cilia classification, even though more precise, is complicated by significant phenotypic variability and clinical or pathologic overlap among various mutation-defined entities (
5). Furthermore, the biologic function of cystic disease-related mutations is not fully understood, even though the frequently observed multiorgan involvement in hereditary cystic kidney diseases is far better explained, because cilia are widespread in tissues; thus, mutations in ciliary genes may affect multiple organs (
3). Considering that a molecular/genetic classification of cystic kidney diseases is a work in progress and no universally accepted classification currently exists, the classification in the previous edition of this book still is valid and therefore is used in this edition with only minor modifications (
Table 4.1). However, the many discoveries about the genetic basis for cystic diseases are reviewed in the context of this classification.
In examining cystic kidneys, the gross morphology and radiologic appearance are very important. The following basic questions are a useful guide: (a) are the cysts bilateral or unilateral, (b) focal or diffuse? or (c) is the kidney enlarged or approximately normal size? Multiple bilateral renal cysts are frequently hereditary, in contrast to isolated, unilateral cysts that are most often acquired. The term “polycystic” is conventionally reserved for ADPKD and autosomal recessive polycystic kidney disease (ARPKD). Multicystic is a term used for a subcategory of renal
dysplasia characterized by diffuse cysts that may grossly mimic ADPKD in early childhood, but the dysplastic features are apparent microscopically. Most cysts are tubular in origin; when cysts involve the glomeruli, cystic dilation of the Bowman space (>twice normal) involving greater than 5% of glomeruli are involved, the diagnosis of glomerulocystic kidney disease (GCKD) is appropriate. GCKD can be primary or secondary; for example, childhood ADPKD may present as primary GCKD and vascular ischemia can cause secondary GCKD (
6).
Medullary cystic disease is an autosomal dominant tubulointerstitial disease of adults with end-stage renal disease (ESRD) occurring in the fourth to seventh decade. Two genetic loci for medullary cystic kidney disease (MCKD) are described: MCKD1 and MCKD2. Symptoms are similar but neither imaging studies nor histopathologic findings are specific as discussed later (
7). Notably, some patients do not have cysts at all. MCKD was once thought to be the same entity as juvenile nephronophthisis (NPHP) due to the overlapping phenotypes (
5). Clearly, NPHP is genetically distinct from MCKD with about 13 different genes causing a variety of clinical NPHP phenotypes. The MCKD1 locus is mapped to chromosome 1q21, but the gene remains unknown. The MCKD2 locus on chromosome 16p12 is identified as the uromodulin (UMOD) gene. UROM mutations may present as GCKD. Therefore, some authors prefer the term uromodulin disorders. Other authors propose to reclassify MCKD as one of the four types of autosomal dominant interstitial kidney disease (ADIKD). UMOD mutations in the proposed ADIKD classification encompass (a) MCKD2, (b) familial juvenile hyperuricemic nephropathy (FJHN), (c) uromodulin-associated kidney disease (UAKD), and (d) GCKD. Once again, we believe that at this time, our classification may be preferable for the intended readership.
Medullary sponge kidney (MSK) is a disease of adults characterized by cysts in the collecting ducts. Cysts are often discovered incidentally by excretory urography in patients investigated for urinary tract infections or stones. On radiology, calcified collecting ducts appear as filling defects (
8). Kidney cysts can be neoplastic (
9). Renal cysts associated with hereditary cancer syndromes such as von Hippel-Lindau (VHL) disease and tuberous sclerosis (TSC) are typically bilateral and multiple. Nonneoplastic, multilocular renal cyst is a distinct tumor mainly of childhood, also called cystic nephroma. The terms multilocular cystic nephroma (MCN), polycystic nephroblastoma, and cystic differentiated nephroblastoma describe the same entity with nephroblastoma added in some neoplasms that mimic Wilms tumor. For example, an intermediate form between multilocular cyst and Wilms tumor is described as cystic partially differentiated nephroblastoma. MCN is typically unilateral; bilateral presentation is exceptionally rare (
10). A new association of pediatric cystic nephroma with lung tumors such as pleuropulmonary blastoma was recently reported; some patients have familial disease, suggesting that cystic nephroma that was once considered a benign and sporadic tumor at least in part has a genetic basis (
11). An entity called localized cystic disease of the kidney is also described, apparently with no hereditary basis (
12).
Simple cortical cysts, extrarenal cysts (parapelvic lymphangiectasis and perinephric pseudocysts), and hemodialysis-induced cysts have distinct pathogeneses and are discussed as separate entities (
Table 4.1).
Polycystic Kidney Disease
Autosomal Dominant Polycystic Kidney Disease
INCIDENCE, CLINICAL PRESENTATION, AND GENETICS
ADPKD is the most common inherited cystic kidney disease affecting 1:500 to 1000 individuals worldwide and 2.3% of patients on dialysis in the United States (
13,
14,
15).
Both men and women are affected, but women have a less severe disease. ADPKD is a systemic disease, and patients
experience multiple renal and extrarenal complications.
Clinical symptoms usually appear after the third decade, but the phenotypic spectrum ranges from in utero onset to adequate renal function at old age. Common complications include kidney stones, infections, flank pain, gross hematuria, and hypertension. Hypertension and gross hematuria are often diagnosed before the age of 30 and thought of as markers of progressive disease (
15,
16). ESRD is common with mean age at 59 years, while approximately 15% of ADPKD patients retain adequate renal function at 80 years (
17). Family history of ESRD plays an important role in predicting ESRD. ESRD at an earlier age (before 55 years) in at least one family member correlates with PKD1 mutations; if at least one family member developed ESRD greater than 70 years of age, this family history was predictive of PKD2 with a 100% predictive value (
18). The liver is involved in 94% of cases in the Consortium of Radiologic Imaging Studies of Polycystic kidney Disease (CRISP) study and eventually becomes palpable but is unusual to manifest as massive liver disease that requires surgical intervention. A separate polycystic liver disease without renal involvement exists and is caused by mutations in non-ADPKD genes as discussed later under polycystic liver disease. Women have different rates of occurrence of renal and extrarenal complications in ADPKD, including liver cyst growth and expansion. Women tend to have more severe liver disease stimulated by estrogens (
17,
19). Women develop renal insufficiency with smaller renal volume than men, whereas liver cystic disease occurs earlier and more frequently in women than in men (
Table 4.1). Pancreatic cysts are rare (present in only around 10% of patients), which is a distinguishing feature from other multiorgan hereditary cysts such as VHL disease (
20,
21). Other extrarenal manifestations include increased incidence of intracranial aneurysms (approximately 8% of ADPKD patients) and extracranial aneurysms, colon diverticula, and mitral valve prolapse.
Genetics have brought unprecedented insights in ADPKD pathogenesis. Ninety percent of affected individuals have a parent with the disease, and their children have a 50% chance of inheriting the condition. De novo mutations may account for about 10% of cases. Mutations are found in two genes, PKD1 and PKD2. A third gene (PKD3) was suspected based on reported isolated cases (less than 1%) but is not yet confirmed. However, lack of evidence in spite of intense search for a third gene is now suggesting that it likely does not exist (
22). Recent methodologic advances have improved mutation detection, and at least 700 PKD1 and over 100 PKD2 mutations are reported, most of which are private (
17,
23,
24,
25). PKD1 contains 4,302 amino acids and PKD2 968 amino acids. PKD1 mutations were thought to affect 80% to 85% of patients, and PKD2 mutations the remaining 10% to 15%, but new data show that PKD2 may be more common affecting 26% to 36% of ADPKD patients (
22). Most mutations are missense and inactivating, but there is no good genotype/phenotype correlation suggesting complex mechanisms including factors that affect protein trafficking (
26), the mutation type on cilia, its functionality, and dosage, factors that may explain the variable clinical manifestations. For example, experimental data show that mutation characteristics may predict ESRD and rate of cyst development/growth in humans. Hopp et al. (
27) show that Pkd1+ mice with PKD1p.R3277C(RC) knockin develop rapidly progressive disease in contrast to Pkd1+/null mice that are normal. This model in which the level of functionality of the mutation product (dosage) determines cystogenesis mimics in utero onset in humans and typical ADPKD presentation in adults, respectively. In addition, concurrent mutation in other genes may also play a role. For example, the protein product of the gene defective in cystic fibrosis (CF), cystic fibrosis transmembrane conductance regulator (CFTR), plays a crucial role in fluid accumulation promoting cyst swelling (
28). Patients with PKD1 have earlier onset of symptoms, in contrast to patients carrying PKD2 mutations who tend to have delayed cyst formation, hypertension, and ESRD, by 10 to 20 years. Both PKD1 and PKD2 genes exhibit extensive allelic heterogeneity that correlates with variable clinical manifestations (
Table 4.2); 18% to 59% of the phenotypic differences in PKD1 are attributed to inherited background genes and perhaps environmental factors that modify disease expression but have no significant difference in life expectancy.
The PKD1 gene is located on chromosome 16p13.3 and consists of a large 53-kb genomic DNA with 46 exons. It encodes a 460-kDa protein, polycystin 1. PKD2 maps to chromosome 4q13-q23 and consists of a smaller DNA sequence of 5.4 kb with 15 exons that encodes polycystin 2. The gene products are called polycystin 1 and 2, respectively. The precise role of mutated polycystins in cyst formation is still under investigation. It is proposed that individual cystogenesis requires biallelic inactivation of a polycystic disease gene through germ-line and somatic mutations within an epithelial cell (the second-hit hypothesis). In this model, the second hit is the limiting step. This hypothesis is supported by genetic studies of cystic epithelia from ADPKD patients and mouse models (
25,
29). Activated normal polycystin 1 is thought to be an epithelial cell membrane receptor sensing cues in the extracellular environment required for renal tubular epithelial cell division and differentiation. Mutated polycystin 1 is only detected in cytoplasmic pools in cystic cells. Polycystin 2 is a smaller molecule localized in the plasma membrane and the endoplasmic reticulum and has structural similarities with a family of sodium/calcium channels, thus thought to modulate intracellular levels of Ca
2+ (
30). Polycystin 1 and 2 have similar cellular distribution providing a biochemical basis for the identical phenotypes caused by PKD1 and PKD2 mutations.
However, other factors such as altered responsiveness to c-AMP may be more important inducers of cysts (
31), and there may be an important role for effects on primary cilia (
25).
Genetic testing is possible by either linkage analysis in large families with several affected members or direct mutation screening. Linkage analysis is limited by the fact that a minimum of four affected family members are required; in small families with a single individual member affected, this test cannot be performed. Direct sequencing is more sensitive and detects about 70% of ADPKD mutations (
32). However, for various reasons in the remaining 30%, the results are ambiguous or negative. Therefore, this approach also has limitations, and there are patients subjected to genetic analysis without definitive results. Genetic analysis methods continue to evolve and include next-generation technologies. Preimplantation diagnosis is also possible (
33).
RADIOLOGIC EVALUATION
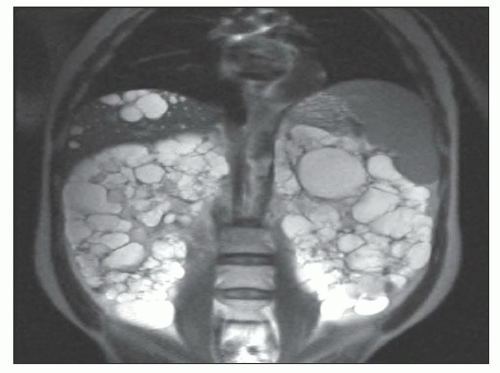
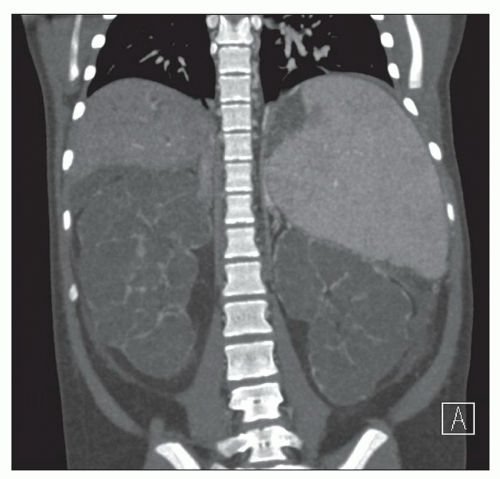
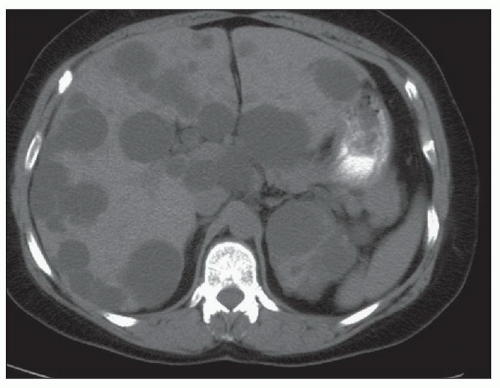
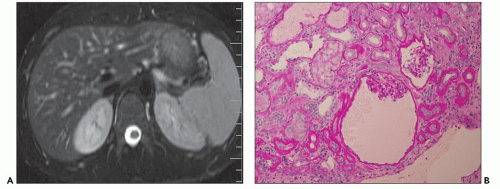
Ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI) are essential for ADPKD diagnosis, patient follow-up, and lately a choice for therapeutic intervention (interventional radiology to minimize cyst volume). Ultrasound is more frequently used for screening, but CT and MRI are more sensitive particularly in early detection of ADPKD when the cysts may be small and or few. Characteristic findings of classic ADPKD are bilaterally enlarged kidneys with multiple cysts with varying signal intensity (
Fig. 4.1). Hemorrhage, tumors within a cyst, or infection generates a high-intensity signal on CT or MRI. Concurrent liver cysts are a frequent finding. Total kidney volume is a major indicator for disease progression, and new methods that reliably measure ADPKD progression were initiated by the CRISP (
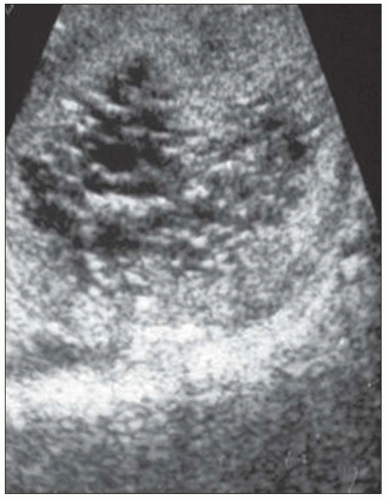
20). The CRISP study revealed that cyst enlargement contributed to renal impairment and correlated with hypertension; in contrast, smaller cysts on MRI were frequently found in normotensive patients. By 30 years of age, most ADPKD patients have radiologic evidence of kidney cysts. Adults less than 30 years of age and children may have asymmetric kidney involvement (unilateral and few cysts). Diagnosis is not problematic in classic adult ADPKD, but phenotypic variability in early stages may hamper recognition and management of patients with only a few cysts, even when family history of ADPKD is known. Ultrasound lacks diagnostic accuracy particularly in infantile and childhood-onset ADPKD. On ultrasound, ADPKD mimics ARPKD and GCK, bilateral cystic dysplasia, and Meckel-Gruber syndrome (
Fig. 4.2). Furthermore, the majority of fetuses screened by ultrasound show no apparent renal or extrarenal cysts, and children may be asymptomatic and not develop cysts for 10 to 20 years after birth. Ninety percent of patients have an affected parent, and diagnosis is highly likely if more than one generation has multiple large cysts with no other symptoms/signs. The traditional guide for family screening is that around 95% of affected individuals will have cysts by the time they reach 30, but Pei et al. (
34) recently refined this algorithm because it tended to underdiagnose PKD2-affected families: three or more renal cysts establish a diagnosis of PKD between 15 and 39 years; two or more cysts in each kidney are sufficient for individuals aged 40 to 59 years; and four or more cysts in each kidney are required for individuals ≥60 years old (
Table 4.3) (
15,
22,
34). Nonetheless, a 19% to 38% false-negative rate is reported in age-specific radiologic analysis of patients with known ADPKD
genotype (
35). This does not include the small proportion (5% to 10%) of children with no apparent history of ADPKD at the time of first presentation and a presumed new mutation. The differential diagnosis includes ARPKD and malignancy, as discussed further below.
PATHOLOGY
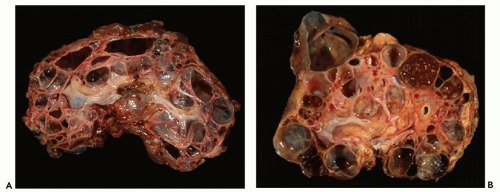
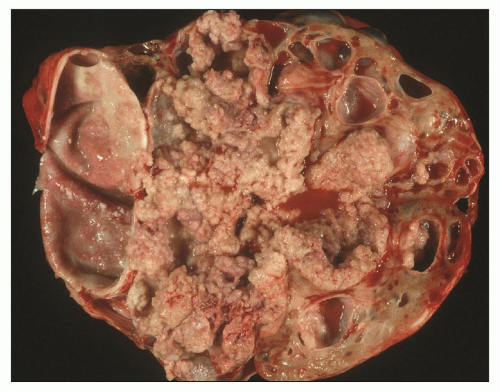
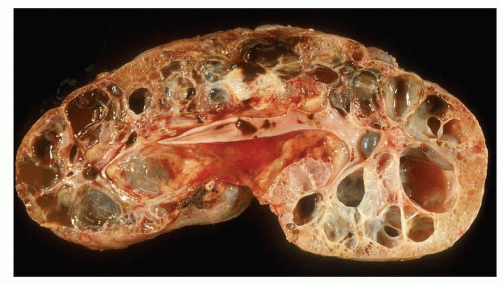
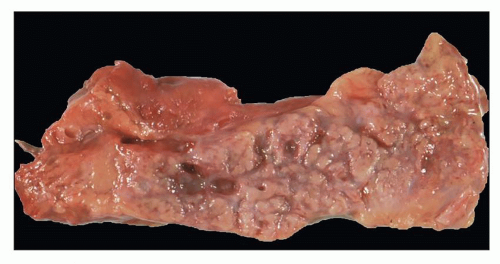
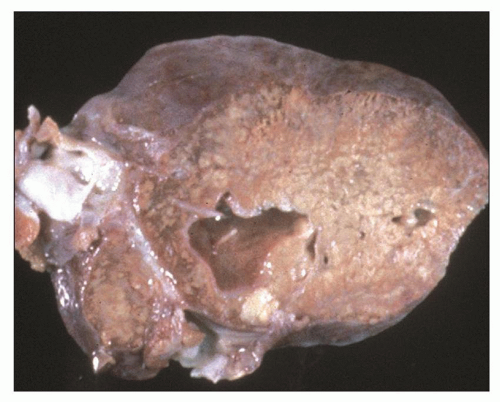
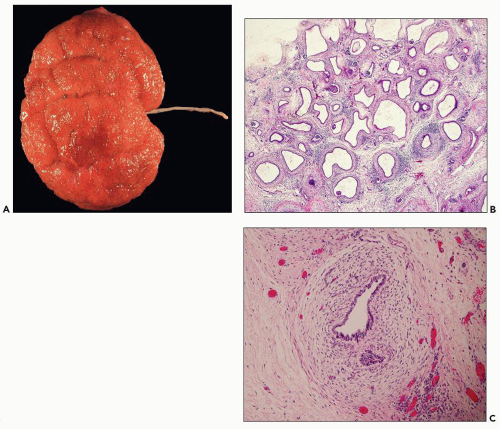
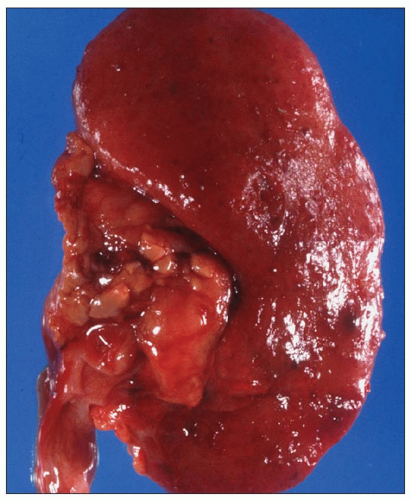
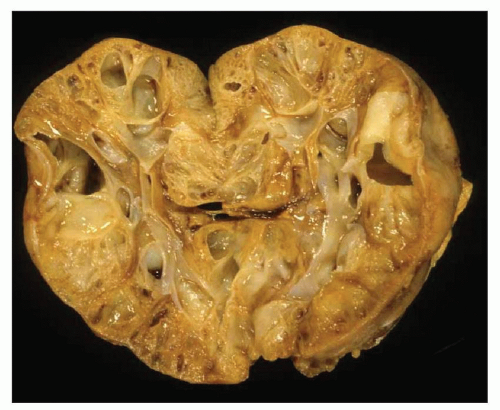
Pathology of ADPKD can be divided into the typical adult presentation and atypical presentation in children (early-onset ADPKD). In adults, ADPKD kidneys are not routinely resected; when patients develop severe complications, such as recurrent and severe back pain, bleeding, or infection, or prior to renal transplantation, surgical removal reveals enormous bosselated kidneys. The kidneys weigh on average 2.5 kg each (mean normal kidney weight equals 0.150 kg). The smaller ADPKD kidneys in adults weighing less than 1 kg may represent PKD2 mutations (see
Table 4.2 and
Fig. 4.3).
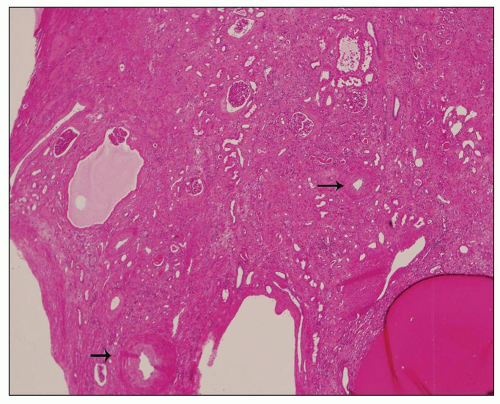
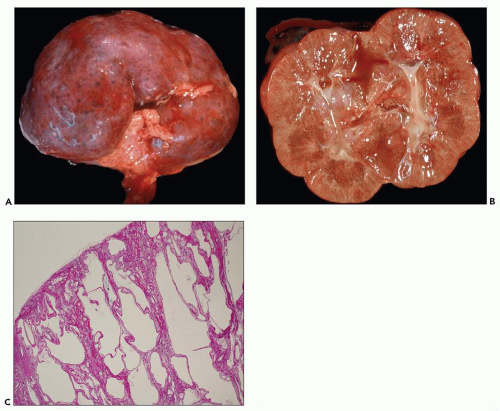
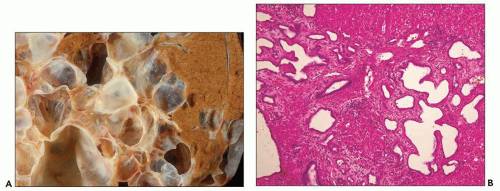
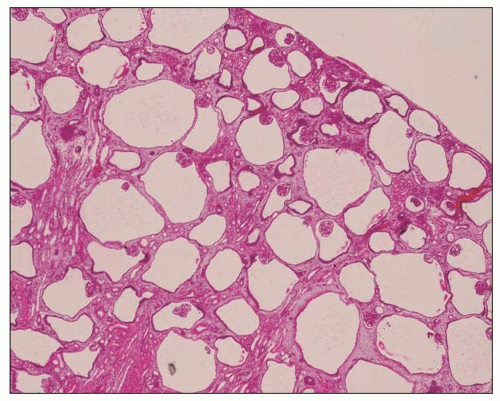
The reniform appearance of the kidneys is lost, and the kidney is distorted by multiple cysts that may contain clear, turbid, gelatinous, or hemorrhagic fluid. On sagittal sections, cysts are typically unilocular, oval, or spherical, vary in size from millimeters to several centimeters, and are randomly distributed. The renal pelvis and calyces cannot be identified, and replacement of the normal renal parenchyma is usually extensive. Residual renal parenchyma is compressed and eventually becomes atrophic by the enlarging cysts filled with eosinophilic fluid (
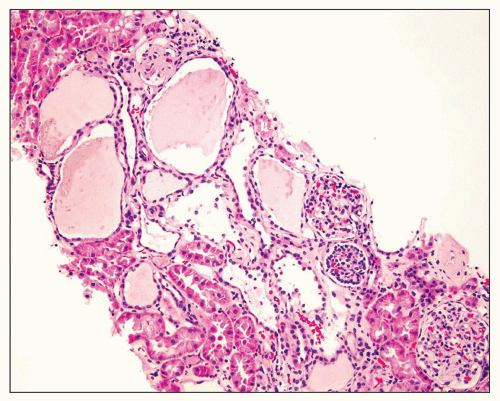
Fig. 4.4). Interstitial fibrosis abounds in most specimens. Globally sclerosed glomeruli are increased; cystic glomeruli are also frequently present likely secondary to vascular ischemia as parenchymal arteries are thickened (
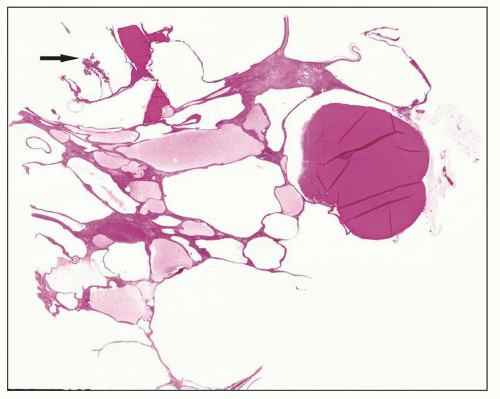
Fig. 4.5). However, even at end stage, there is significant number of intact-appearing glomeruli, which may explain why these severely distorted kidneys may continue to function for a long time in spite of cyst expansion. The epithelium lining the cysts is denuded, flat, or hyperproliferative. Proliferating epithelium within ADPKD
cysts is indeed characteristic and not infrequently gives rise to papillary microadenomas that increased the risk for malignancy (
Figs. 4.4 and
4.6). Micropolyps are present in as many as 90% of patients with ADPKD (
36). They may be small with broad base or flower-like microadenomas.
ADPKD cysts arise in a small fraction (1% to 2%) of proximal and distal tubules early in childhood, and new cysts are unlikely to develop over a patient’s lifetime. Cyst development follows gradual luminal dilation by tubular epithelial cell proliferation, apoptosis, and fluid accumulation. Eventually, cysts separate from the parent tubules and become a sac-like structure while their lining epithelial cells continue autonomous proliferation. Another feature of ADPKD epithelia is proliferation and loss of polarity and show characteristic mispolarization of proteins such as Na
+-K
+-ATPase (
37). Cell proliferation is accompanied by secretion of high amounts of electrolyte transport proteins that result in excessive secretion of solute and fluid into the cysts. These phenomena are consistent with a neoplastic-like phenotype in ADPKD epithelia, perhaps because the mutated polycystins are unable to maintain the normal state of epithelial differentiation and maturation (see under molecular biology of ADPKD). Other factors such as epidermal growth factor may also mediate cell proliferation and fluid secretion within the cysts (
38). The observation that cyst formation involves a fraction and not all nephrons has immediate clinical implications and prompted intense research in designing clinical trials for early intervention to control cyst size (see
Clinical Management, Prognosis, and Therapy). The relationship between kidney size and progression to renal failure is now clearly elucidated. In patients who develop renal failure, there is loss of noncystic parenchyma in association with nephron mass replacement by interstitial fibrosis. In fact, the size of the cysts correlates directly with the loss of renal function. Cysts prevent the drainage of urine from upstream tributaries, cause tubular atrophy, and release chemokines, cytokines, and growth factors resulting in the progression to fibrosis (
39). Finally, while there is a lot of experimental evidence that implicates cilia in ADPKD, abnormal cilia in human disease is rarely demonstrated.
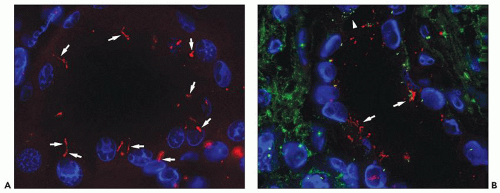
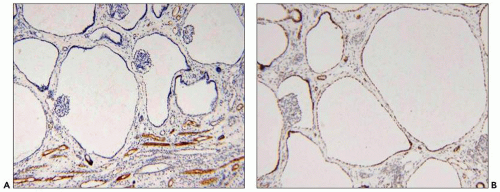
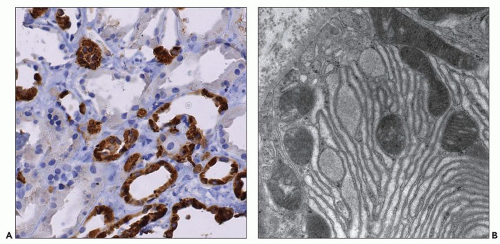
Figure 4.7 shows cilia in normal and ADPKD tubular epithelial cells. Cells lining ADPKD cysts exhibit increased number of cilia and also supernumerary basal bodies compared to normal epithelial cells. These findings are similar to tubular epithelial spheroid cell cultures grown on Matrigel that form hollow structures mimicking tubule formation (
40). Extra cilia derived from the same ciliary pocket (a specialized space in the root of cilia) suggest that the ciliary pocket may be the rate-limiting structure for trafficking of ciliary proteins in human ADPKD (see under Molecular Pathogenesis). However, additional studies in human tissues are required to derive definitive results on the pathology of cilia in human ADPKD.
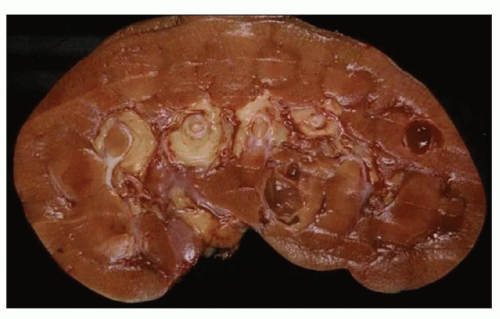
MALIGNANCY IN ADPKD
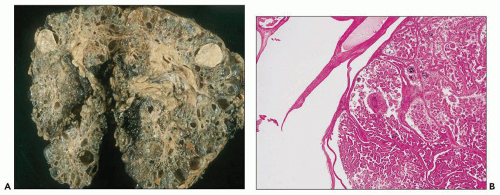
There are numerous case reports of renal cell carcinoma (RCC) developing in ADPKD cystic kidneys and small patient series that show more than 10-fold increase of malignancy compared to the general population. Multifocal and bilateral RCC was reported by Jay Bernstein in as many as 20% of ADPKD kidneys enforcing the view that epithelial cells lining ADPKD cysts have neoplastic properties (
41). However, others find no significantly increased risk. In the last 10 years, we have seen 6 RCCs in 22 resected ADPKD kidneys (that is equal to about 27% of resected ADPKD kidneys), but this number certainly represents a selection bias. Intracystic RCCs usually range from 1 to 4 cm and can be unilateral or bilateral.
Histologically, tumors are typically RCCs, clear cell, papillary, or chromophobe type (
Fig. 4.8). The predominant histologic type appears to be papillary RCC. Tumors are diagnosed on routine radiologic follow-up screening. Beyond RCC, other kidney malignancies, for example, transitional cell carcinoma, are found incidentally (
Fig. 4.9).
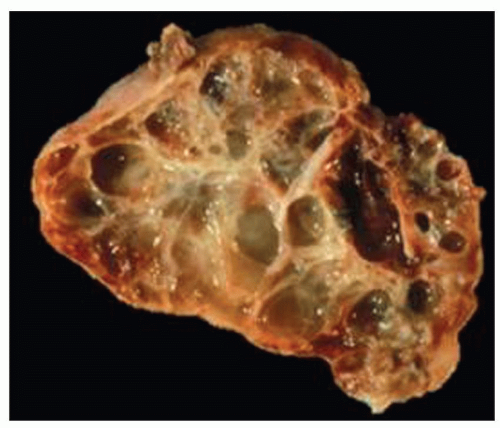
EARLY-ONSET ADPKD
Early-onset ADPKD may start in utero and be apparent on fetal ultrasound. Postnatally, there is a spectrum of lesions ranging from unilateral cystic disease (
Fig. 4.10) to just a few
isolated cysts (
Fig. 4.11). This variability in ADPKD cyst development has prompted a revision of the previously established radiologic criteria for childhood ADPKD that lowered the threshold for at-risk individuals (
34). A rare example of infantile ADPKD with gross pathology similar to adults is shown in
Figure 4.12; cysts are filled with gelatinous fluid, and compared to
Figure 4.3, this infantile kidney is nothing but a
miniature image of adult ADPKD. In childhood ADPKD, concurrent liver or pancreas cysts facilitate the diagnosis (
Fig. 4.13). However, more frequently than one may expect, a renal biopsy is performed to rule out early-onset ADPKD following ambiguous MRI or CT. Such cases also have unclear ADPKD family history and absent extrarenal cysts. A constant and helpful finding is the finding of bilaterally enlarged kidneys (always a good indication for hereditary disease). The example shown in
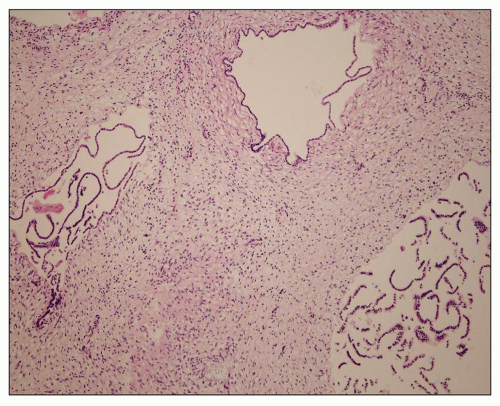
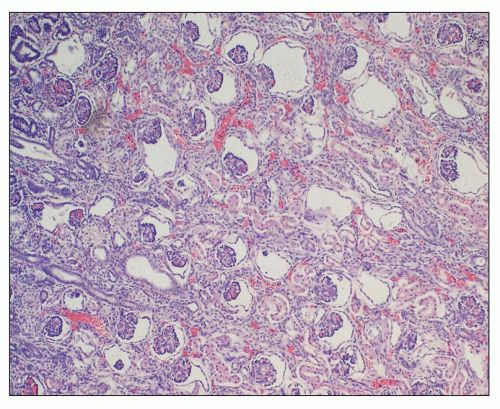
Figure 4.14 is from a 4.5-year-old boy who presented with acute renal failure, fever, and acute pyelonephritis. There was no family history of ADPKD. CT showed an infiltrating pattern suggestive of bilateral nephroblastomatosis or Wilms tumor. There were no liver lesions. Renal biopsy revealed cystic glomeruli and focal tubular dilation, but no evidence of blastema (
Fig. 4.14B). There were no papillary proliferations. Tubular atrophy and chronic interstitial inflammation was minimal. Acute inflammatory cells were present in some
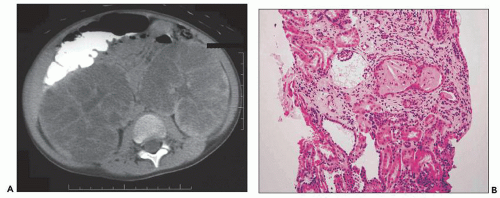
tubules consistent with the recent history of acute pyelonephritis. Features such as in this biopsy may be interpreted as nonspecific, but glomerular cysts in the renal biopsy of a young child with bilaterally enlarged kidneys raise the possibility of early-onset ADPKD to be confirmed with genetic testing. The differential diagnosis of glomerular cysts collectively known as GCKD includes ARPKD (discussed below). Genetic testing is helpful in the majority (greater than 70%) but not all cases. For example, in
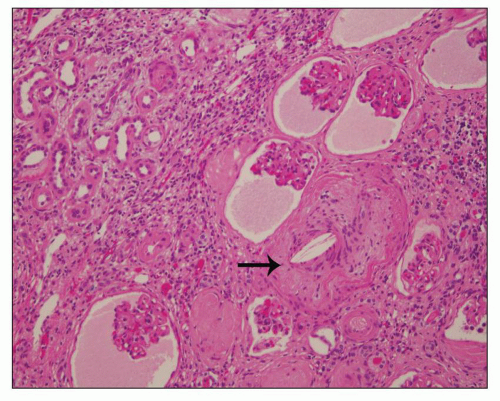
Figure 4.15 is a renal biopsy from a 7-year-old boy with no family history of ADPKD who presented with bilaterally enlarged kidneys and liver cysts radiologically thought to be polycystic kidney disease (PKD). Genetic testing was performed prior to renal biopsy, but the results were ambiguous. The biopsy shows numerous tubular cysts some filled with pale fluid; cysts are oval and focally elongated. A few glomeruli were globally sclerosed, and others were immature (fetal glomeruli). There were no micropapillae. Given the history of liver cysts and in the exclusion of ARPKD, early-onset ADPKD was favored, perhaps due to de novo mutation.
Early-onset ADPKD is not only difficult to diagnose but also difficult to explain with the two-hit hypothesis that presupposes a time interval for the second hit. A “
third-hit signaling” that is thought to promote aberrant cellular proliferation and kidney growth may in part explain the phenotypic variability including early-onset ADPKD (
42). In addition, some patients may have concurrent mutations in other genes, for example, the CF gene (
43), HNFβ1 (
26), or TSC. Modifier genes that may accentuate the disease phenotype without a second hit remain a possibility to explain the presentation in young patients with early-onset ADPKD.
Autosomal Recessive Polycystic Kidney Disease
INCIDENCE, CLINICAL PRESENTATION, AND GENETICS
ARPKD is much less common compared to ADPKD with an incidence of 1:20,000 live births. Typically, ARPKD starts in utero and presents at birth with large kidneys and liver fibrosis. Atypical ARPKD manifests in older children or young adults presenting predominantly with liver disease. About 50% of ARPKD is
perinatal; kidneys are echogenic, and babies develop oligohydramnios leading to lung hypoplasia and death in about 30% of cases. Bile duct dilation and cirrhosis complicated by sepsis are common immediate causes of neonatal death. In older children, liver complications also predominate. A database with clinical information of patients surviving infancy has accumulated data from 34 centers in America (
46). More than 200 enrolled patients were categorized into two groups; 79.4% were born before 1990 and 20.35% after 1990; 85% were alive at 1 month, 78% at 1 year, and 75% at 5 years. Respiratory and chronic renal insufficiency was a significant predictor for mortality. Hypertension and liver disease did not affect survival. The most frequent complications were chronic lung disease, growth retardation, hyponatremia, and urinary tract infections. Hypertension was common but not seen in all patients. Ultrasound evaluation among 191 of the patients in the database, in about 50%, revealed echogenic kidneys without cysts (because masses of tiny cysts increase echogenicity but each individual cyst is below the limits of ultrasound resolution); the remaining had small cortical cysts. Liver imaging was normal in about 50% of patients; 16% had liver cysts consistent with Carol disease and of those 3 (13%) had episodes of acute cholangitis. These data and additional studies show that patients who survive infancy do much better than previously reported with a mean life expectancy of 27 years (range 18 to 55) (
47). A minority of patients will not progress to ESRD, but most will have dialysis or transplantation. Complications may arise in women during pregnancy, such as acute decline of renal function and preeclampsia (
48).
In ARPKD, neither parent has the disease; each child of parents who are both carriers has a 1:4 chance of inheriting the disease and 1:2 chance of being a carrier. All patients with ARPKD have mutations in the polycystic kidney and hepatic disease 1 gene (PKHD1), which is located on chromosome 6p21 and encodes a protein named fibrocystin/polyductin; greater than 300 mutations were identified in neonatal disease. There have been no new genes identified to explain infantile versus juvenile onset. Specific genotypes according to some authors affect the severity of renal and hepatic histologic abnormalities. For example, a study compared 54 fetuses with medical pregnancy termination to 20 neonates who died shortly after birth and found 55.5% of the mutations were truncated fibrocystin. Presence of two truncating mutations correlated with the most severe prenatal ARPKD (
49). However, a different study of neonatal survivors (78 children and adults) identified 77 mutations (41 new), 19 of which were truncating in spite of postnatal onset (
50,
51). Haplotype-based diagnostic tests for at-risk pregnancies are available with a mutation detection rate of about 60% to 80% with those with kidney cysts and about 30% for those with liver disease but no renal cysts. In 12/78 cases, the criteria for ARPKD were not met, and GCKD, ADPKD, and other entities were thought possible but not confirmed. In spite of some limitations of genetic testing, preimplantation genetic methods are helpful and constantly improving (
52).
RADIOLOGIC EVALUATION
Neonatal ARPKD is diagnosed by fetal ultrasound as early as 17 weeks of gestation. Kidneys are bilaterally enlarged and hyperechogenic, echogenicity attributed to innumerable 1- to 2-mm cysts in the collecting ducts that increase acoustic interface frequencies. MRI can be used for the evaluation of fetuses that have equivocal second-trimester ultrasound. In the last few decades, ARPKD occurring in older children is recognized (
50). Findings range from bile duct dilatation to portal hypertension to esophageal varices and enlarged liver and spleen.
Figures 4.16 and
4.17 are from a 24- and a 23-year-old woman, respectively. Ultrasound shows enlarged cystic kidneys, and CT clearly demonstrates concurrent kidney enlargement, liver cirrhosis, and splenomegaly. Liver and kidney dysfunction appear independent in late-onset ARPKD; patients may present with liver symptoms first, and they may reach middle age without ESRD. Atypical cases with no liver lesions may be mistaken for ADPKD or malignancy.
PATHOLOGY
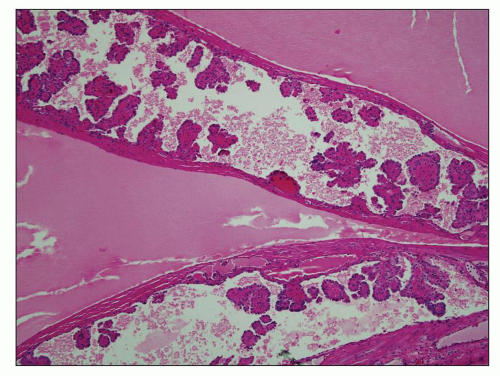
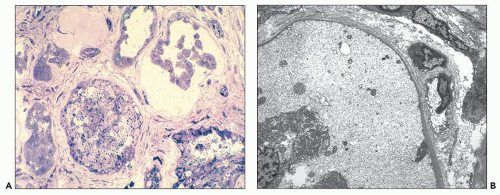
Neonatal ARPKD typically presents with congenital kidney enlargement and liver fibrosis. The kidneys show diffuse microcysts, which give these kidneys a sponge appearance (
Fig. 4.4.18A). Sagittal sectioning reveals diffuse replacement of the renal parenchyma by cylindrical cysts (
Fig. 4.18B). Microscopically, cysts involve the collecting ducts and typically extent to the cortex (
Fig. 4.18C). There is no increased interstitial tissue. Autopsy studies showed that collecting duct dilatation and renal cortical involvement increase with gestational age (
49). However, cysts may be entirely absent in early gestation, focally present in midgestation, or involving the kidney globally in late gestation. Type of mutation also affects the extent and time of cyst development. For example, in the study by Denamur et al., two severe (truncating) ARPKD mutations correlated with diffuse cylindrical cysts. Degenerating kidney cysts may present diagnostic difficulties as cysts may appear irregular with a branching pattern instead of cylindrical (
Fig. 4.19). Histologic variability prompted some authors to devise a scoring system that takes into account the extent of collecting duct dilation, loss of proximal tubules, cortical involvement, and loss of the nephrogenic zone; four grades 1 to 4 were proposed. Based on this scoring, ARPKD in
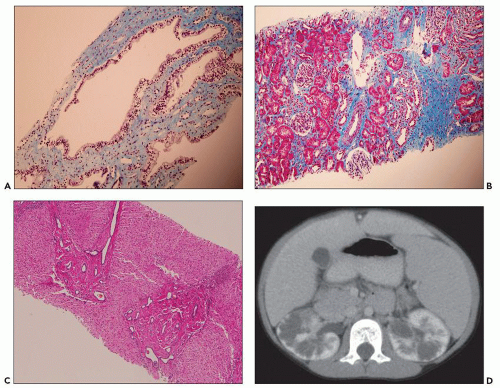
Figure 4.18 represents a grade 3 lesion. In the renal biopsy submitted from a 7-year-old boy with clinically unexpected ARPKD, we found medullary duct ectasia with normal lining and intact renal cortex; there were no typical cylindrical cysts extending into the cortex (Denamur grade 2) (
Fig. 4.20A and B). This scoring system may not be applicable in all cases (e.g.,
Fig. 4.19) but raises awareness of the subtle findings in ARPKD and indicates that absence of typical long cylindrical cysts does not exclude ARPKD diagnosis. Liver biopsy was also performed in this 7-year-old boy and showed bile ductile proliferation (
Fig. 4.20C). MRI imaging revealed an ambiguous infiltrative pattern suggestive of bilateral lymphoma (
Fig. 4.20D).
The differential diagnosis of neonatal ARPKD includes early-onset ADPKD and bilateral cystic renal dysplasia sporadic or syndromic and Meckel-Gruber syndrome. Meckel syndrome is an autosomal recessive disease characterized by cystic kidneys associated with hepatic fibrosis, polydactyly, and central nervous system abnormalities such as encephaloceles. Both the kidneys are enlarged, but microscopically, the kidneys are dysplastic.
Polycystic Liver Disease and Hepatic Fibrosis
Polycystic liver disease encompasses three etiologies: (a) cysts associated with ADPKD, (b) a separate entity of autosomal dominant polycystic liver disease (ADPLD) that is not accompanied by kidney cysts, and (c) Caroli disease. The most frequent is extrarenal ADPKD. Liver cysts may cause severe liver dysfunction when they are multiple occupying a large liver segment (
Fig. 4.21), but more frequently are focal and asymptomatic. Polycystic liver disease is arbitrarily defined as a liver that contains greater than 20 cysts (
53,
54,
55). Cysts are intrahepatic or perihilar replacing the liver parenchyma (
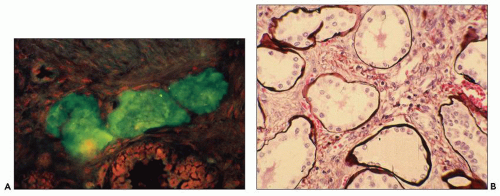
Fig. 4.22A). Microscopically, cysts appear disconnected from the biliary tree and composed of dilated bile ducts embedded in a fibrous stroma (known as von Meyenburg complex) (
Fig. 4.22B). These are thought to derive from abnormal ductal plate formation in utero justifying the term biliary dysgenesis. In ARPKD, there is also abnormal formation of bile ducts and defective apoptosis of excess precursor cells leading to increased number of bile ducts, abnormal brunching, and invariably periportal hepatic fibrosis (
Fig. 4.23). Septic cholangitis and death from septicemia is a severe complication in neonates. Rarely, hepatic fibrosis instead of liver cysts may be seen in patients with ADPKD and or liver cysts are reported in children with ARPKD (
56,
57). These exceptions to the more typical kidney-liver phenotypes raise questions about the mechanisms of such crossover presentations, for example, the role of epithelial stromal interactions and or background gene modifiers. Malignancy is rare but at least one case of cholangiocarcinoma developing in polycystic liver disease in an adult is reported (
53).
ADPLD is a heterogeneous entity; 80% of patients presumed to have ADPLD have no detestable gene mutations.
A sizable minority has mutations in two genes: one on the short arm of chromosome 19 affecting protein kinase substrate 80K-H (PRKCSH) coding for a protein called hepatocystin and a second on chromosome 6 involves Sec63. These proteins localize in the endoplasmic reticulum of cholangiocytes and are thought to mediate cell proliferation and fluid secretion via increased 3′,5′-cyclic adenosine monophosphate (cAMP). Evidence derives from studies that show secretin (a cAMP agonist) to increase fluid production in hepatic cysts and somatostatin analogs to suppress the size of liver cysts (
58,
59,
60). These studies brought forth new compounds as therapeutic agents targeting reduction of cell proliferation and fluid secretion by liver cysts.
PATHOGENESIS
The pathogenesis of liver cysts is thought to be due to persistence or lack of remodeling of the embryonic ductal plate, which normally takes place during embryogenesis (
61). Hepatic precursor cells normally migrate from the foregut and form a double layer around the portal veins, which is called the ductal plate. The ductal plate remodels into bile ducts and portal tracts over several weeks during embryonal development. The rest of the precursor cells undergo apoptosis. If precursor cells do not involute or do not respond to proper signals and remain unincorporated, they may lead to ductal plate dysgenesis. It is interesting that among other factors participating in biliary morphogenesis, liver-specific loss of HNF1β produces immature ductal plates and loss of planar cell polarity of cholangiocytes (
61). Other investigators have studied cilia of cholangiocytes and gene mutations that impair function of cilia. Cilia in small liver cysts (less than 1 cm) appear normal; larger cysts greater than 1 cm have shortened cilia, while larger cysts (greater than 3 cm) show no cilia. In addition, cyst epithelia show increased cell proliferation and increased expression of estrogen receptors, IGF1, IGF1-R, proliferating and pAKT (
62). Furthermore, some patients with HNF1β are reported to
lack cilia on cholangiocytes (
63). These structural and functional ciliary defects are somewhat similar to ciliary dysfunction in kidney cyst formation.
Clinical Management, Prognosis, and Therapy
Recognition of treatable complications of ADPKD in recent decades facilitated early diagnosis and better management. Fast kidney enlargement detected by ultrasound occurs in some but not all individuals. Those with slower renal growth have less associated risk factors such as hypertension and decreased glomerular filtration rate. Progressive disease manifests with increased overall kidney and individual cyst size; therefore, new therapies target minimizing cyst size and renal growth by selective surgery (
64) or with experimental drugs that exploit new molecules as revealed by clinical trials and animal studies. The latest is Tolvaptan (vasopressin V2-receptor antagonist), which has a modest effect in slowing renal failure and kidney size (
65). Renal transplantation is also a choice. Treatment of liver disease is similarly multifaceted and includes partial liver resection, transplantation, and/or experimental drugs (
15).
Glomerulocystic Kidney Disease
GCKD is not one disease but a heterogeneous group of entities with glomerular cysts as the common histologic finding (
Table 4.4). By definition greater than 5% of glomeruli should be cystic. A glomerular cyst is defined by a dilated Bowman space greater than or equal to two times the normal (
6,
66). GCKD can be classified into primary and secondary types. Primary GCKD includes GCKD associated with heritable diseases such as (a) ADPKD, (b) ARPKD, (c) ADGCKD due to UROM, HNF1β, and renin mutations; (d) TSC, and (e) NPHP. Secondary GCKD includes (a) renal dysplasia, sporadic/nonhereditary, obstructive, and syndromic and (b) ischemic GCKD. Kidneys may be enlarged, normal in size or small. ADGCKD is clinically heterogeneous and includes families that have mild chronic disease with hypoplastic kidneys in some individuals, or normal size kidneys in others,
and separate families characterized by maturity-onset diabetes of the young (less than 25 years of age) (MODY) and renal cysts (
67). ADGCKD with normal or smaller than normal kidneys was found in an Italian family with glomerular cysts, no extrarenal cysts, and an overall benign course with stable renal failure (
68). A missense mutation (c315R) in the gene for uromodulin (UMOD), also known as Tamm-Horsfall protein, was found in some family members (
69). Subsequently, a separate family with ADGCKD and UMOD mutations was reported independently (
70). Interestingly, all members of this unique family had severe impairment of urine-concentrating ability and hyperuricemia resembling the phenotype of NPHMCDK complex and severe reduction of excreted uromodulin. Immunohistochemistry with antibodies to uromodulin revealed dense intracellular accumulation of uromodulin in tubular epithelia of the thick ascending limb of Henle loop in kidney biopsies. Confusion exists in the literature when it comes to the specific renal biopsy findings of patients with reported UMOD mutations because there is no microscopic description in all cases, and some authors prefer to categorize UROM mutations according to presenting symptoms, for example, hyperuricemia, or consider these in the spectrum of tubulointerstitial nephritis and NPHP. UMOD mutations are responsible for three autosomal dominant tubulointerstitial nephropathies including medullary cystic kidney disease type 2
(MCKD2), FJHN, and GCKD. Nonetheless, it appears that most patients with UMOD mutations do not have glomerular cysts, and there are patients with hyperuricemia and glomerular cysts who upon genetic analysis show no evidence of UROM mutations. In MODY, hepatocyte nuclear factor (HNF) 1β mutations in a fraction of patients cause early-onset type II diabetes and kidney cysts. Diabetes is overt in about half of the patients with documented mutations. In others, diabetes is found upon screening (
71). Some patients have glomerular cysts, others have small kidneys with oligomeganephronia (few but large glomeruli), or renal dysplasia and anomalies of the lower genitourinary tract. The reasons for phenotypic variability in MODY remain unclear. However, HNF1β plays an important role in nephron and biliary development in animal models and is a regulator of UROM transcription. Both HNF1β and mutated UROM are implicated in abnormal cilia formation in human ADPKD/ARPKD (
72,
73).
PATHOGENESIS
Understanding of cystic renal disease has greatly advanced in the past few decades. However, the exact pathogenesis of GCKD is still unclear. At least four proposed mechanisms to explain glomerular cyst formation have been proposed: (a) intrarenal medullary inflammation or intrarenal medullary obstruction during the last 10 weeks of gestation, (b) altered collagen composition of the Bowman capsule, (c) stenosis at the glomerulotubular junction, and (d) ciliary dysfunction. However, intrarenal obstruction during fetal development does not explain the predominately cortical distribution of cysts; a weak Bowman capsule that would facilitate dilatation of the urinary space as prourine is formed during kidney development has not been proven, and three-dimensional reconstruction and image analysis have excluded glomerulotubular junction stenosis/obstruction (
6). The cilia hypothesis is currently the prevailing one to explain ADPKD/ARPKD and other hereditary cystic diseases based on data that support tubular epithelial proliferation, fluid accumulation, and remodeling of the extracellular matrix contributing to cyst formation. It is possible that the same factors underlie cyst formation in GCK. It is interesting that developing podocytes do have cilia in contrast to mature podocytes, but there is almost nothing known about their physiologic role or function (
76). Environmental factors (e.g., gestational maternal phenacetin), toxin exposure, infections, or drugs (ego, lithium) have also been postulated. With
the exception of lithium and even though GCKD in humans shows remarkable resemblance to cysts in the rabbit following long-standing corticosteroid treatment, the contribution of steroids in human disease is vague. Among numerous mouse models for cystic kidney diseases, GCKD is evident in 25% of aged +/jcpk heterozygotes (
66). This and other animal models offer an opportunity to study the enigma of GCKD.
Renal Medullary Cysts
Nephronophthisis and Medullary Cystic Kidney Disease
INCIDENCE, CLINICAL PRESENTATION, AND GENETICS
Taken together, the NPHP family of syndromes is the most frequent genetic inherited cause of ESRD in children and adolescents according to some authors (
77). Inheritance is autosomal recessive, and traditional cloning and conditions include the original NPHP group and the associated Senior-Loken, Joubert, Meckel-Gruber, Cogan, and Mainzer-Saldino syndromes (
78). NPHP and associated disorders are considered ciliopathies, as all NPHP gene products are expressed in the primary cilia, similarly to the PKD proteins (
79). These are caused by mutations in over 18 genes, mostly highly conserved across evolution, with a striking common feature that their encoded proteins all appear to localize to the primary cilia or centrosomes. This is not just in renal epithelial cells, but in other affected sites such as the cerebellum, liver and bone, and the retina where rods and cones are modified cilia.
Classical juvenile NPHP is characterized by insidiously progressive tubulointerstitial nephritis that progresses to endstage renal failure usually during adolescence. Increased thirst and urination are the typical first signs at around 3 years of age, secondary to a urinary concentrating defect, followed by failure to thrive and rising creatinine. Kidneys may be relatively normal size but loss of corticomedullary differentiation occurs as the disease progresses and cysts occur at the corticomedullary junction around the time children reach CKD V. NPHP1, 3, 4, 5, 6, 7, 8, and 9 mutations have been detected in juvenile NPHP, while NPHP2 accounts for the rarer infantile form that is characterized by cortical microcysts and end-stage disease before the age of 5 (
79).
Pathways disrupted in various NPHP include canonical and noncanonical Wnt signaling, Sonic hedgehog, and Hippo (
80,
81). Inversin, the gene product of NPHP1, illustrates the complexity and interactions within this group: inversin colocalizes with nephrocystin and β-tubulin in primary cilia (
82) and acts as a molecular switch between different Wnt signaling cascades by inhibiting the canonical Wnt pathway via increased turnover of disheveled, which is downstream of the frizzled wnt receptor, while promoting noncanonical signaling required for convergent extension (at least in
Xenopus) (
83).
MCKD has autosomal dominant inheritance and is relatively rare with an annual incidence of 34 to 56 new cases per year reported at the United States Renal Database (www.usrds.org/request.asap). The main clinical symptoms that NPHP and MCKD share are decreased urine-concentrating capacity, polydipsia and polyuria, and renal cyst formation in the corticomedullary junction or the medulla. An association with hyperuricemia and gout is recognized. However, clinical presentation varies greatly across these groups. For example, polydipsia/polyuria may be mild or absent, and renal cysts may only be found at autopsy and not by ultrasound or CT (
84). Ultrasound detects cysts in only 40% of the patients. Patients with no cysts may not be diagnosed until late in life or at autopsy. Cysts are unilateral or bilateral, and they vary in number. In a large 186 member Cypriot family, bilateral cysts were found in only 12.5% of carriers (
84). Less than half of these patients had hyperuricemia, and clinical gout was only reported in five. Therefore, while clinical symptoms and presence of cysts are part of the disease phenotype, none are sensitive enough for diagnosis. Heredity may not be apparent at first either. There are two genes (and perhaps a third) designated as MCKD1 and MCKD2. ESRD in MCKD1 develops at about 62 years and in MCKD2 at about 32 years. A father-to-son transmission suggests an autosomal dominant mode. Patients often develop hypertension early but, paradoxically, some develop hypotension later due to salt wasting. There are
also associations such as hypogonadism, epilepsy, and spastic quadriparesis among others. A locus on chromosome 1q21 for MCKD 1 was identified in the Cypriot family presenting with very late onset of ESRD. A second locus on chromosome 16p12 for MCKD2 that encodes for uromodulin was found in an Italian family and confirmed by independent investigators in a Welsh family in the United Kingdom (
85,
86). A third MCKD gene is thought possible by Kroiss et al. Familial hyperuricemic nephropathy presenting during childhood was described in two Czech families and one Belgian family (
87). The gene in these families is located close to 16q12 (16q11 locus in fact), suggesting that more loci will be identified in families with a combination of MCDK symptoms and variable ESRD onset.
Currently, hyperuricemia is the common finding in three clinical entities related to UROM gene mutations, also referred to as
uromodulin disorders. These are MCKD2, FJHN, and GCKD. UMOD gene encodes for uromodulin, an 85-kDa glycoprotein involved in renal stone formation and urothelial cytoprotection. Patients often present in early adulthood with hyperuricemia or gout and normal blood pressure. ESRD develops in 15 to 20 years. At least 40 UMOD mutations are reported. Renal biopsy findings in most patients with UROM mutations do not show glomerular cysts but tubulointerstitial fibrosis, a phenotype seen in NPHP/MCKD diseases (
88).
Pathology and Pathogenesis
The pathology of NPHP and MCKD is nonspecific. In juvenile NPHP, the kidneys are grossly normal or slightly decreased in size. Corticomedullary or medullary cysts are present in some patients (
Fig. 4.30). Histopathologic findings consist of a triad of findings: tubular cysts, tubulointerstitial inflammation and fibrosis, and tubular basement membrane disruption. Glomerulosclerosis is invariably present but varies between 20% and 50%. Cystic dilation involves distal tubules and collecting ducts. Dilated tubules may contain acellular material that stains positive with antibodies to uromodulin (also known as Tamm-Horsfall protein). These histopathologic findings are more frequent in juvenile and adolescent NPHP, while infantile NPHP often presents with glomerular cysts. Uromodulin was initially proposed as an NPHP hallmark and the result of an intrinsic tubular defect that allows it to escape into the lumen or the interstitium (
Fig. 4.31A). However, uromodulin accumulation in renal tubules is seen in association with increased intratubular pressure in obstructive and reflux nephropathy in which tubular basement membrane disruption is not the primary defect, as well as chronic nephropathy, stone disease, and other conditions (
73). Nonetheless, disintegration of the tubular basement membrane is an important finding in NPHP-MCKD (
Figs. 4.31B and
4.32A). Electron microscopy reveals dilated tubules with irregular out pouching and tubular basement membrane thickening, splitting, and replication, alternating with thin or entirely absent segments (
Fig. 4.32B). Patients with some NPHP patients with detectable UROM mutations have distinct features on electron and light microscopy. These consist of uromodulin accumulation in the distal tubules (
Fig. 4.33A) and hyperplastic rough endoplasmic reticulum (RER) and amorphous deposits within dilated RER cisternae (
Fig. 4.33B).
Pathogenesis of cyst formation and predilection of the corticomedullary junction in NPHP-MCKD continues to be an enigma, but several hypotheses are now actively explored. Once again the cilia hypothesis predominates and a wealth of information from animal models has brought unprecedented insights into these diseases (
89). For example, inversin is thought to act as a molecular switch between the different Wnt signaling pathways and is associated with the accumulation of proteins within the cilia. Nephrocystin-1 is localized to centrosomes in interphase and the mitotic spindle pole during mitosis, while the product of NPHP9 localizes to the proximal region of the primary cilium and thought to modulate ciliary targeting of polycystin 1 and 2 (
89). In animal models, the mutant forms of uromodulin cause the protein to be retained in the RER, which inhibits normal trafficking and expression at the cell surface. Accumulation within the RER may interfere with appropriate localization of the mutant protein within the physiologic site in the basal aspect of tubular epithelial cells.
Medullary Sponge Kidney
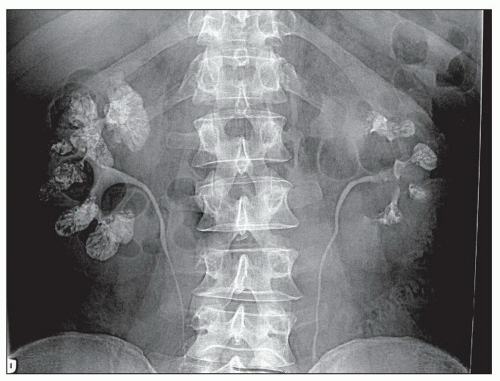
MSK is a cystic disease of adults with cysts that arise in the medullary collecting ducts. Cysts contain numerous calcium deposits or stones and may reach 8 to 10 cm in diameter. Patients present with hematuria, infection of the urinary tract, or obstruction. Upon radiologic investigation, bilateral nephrolithiasis is found in most individuals (
96). A typical example is shown in
Figure 4.35. The excretory ureterogram shows filling defects in the medulla without hydronephrosis. Lithiasis often takes interesting shapes and has been likened to a flower bouquet or “papillary blush” by radiologists.
Unilateral or segmental disease is rare. MSK in the majority of patients is a nonhereditary disorder, but approximately 12% of patients appear to have familial disease. Italian investigators recently reported mutations in the glial cell-derived neurotrophic factor (GDNF), a ligand for RET receptor signaling and genitourinary development. Mutations in GDNF and RET cause urinary tract malformations collectively known as CAKUT (discussed under renal malformations). How GDNF mutations may cause sponge kidney remains to be investigated (
97).
Molecular Pathogenesis of Cystic Kidney Diseases
There has been an explosion in genetic information regarding the mutations underlying diverse cystic kidney diseases since the turn of the century, and the majority appear linked to a
previously underwhelming cellular organelle—the primary cilium. Hence, many apparently distinct conditions have now been ascribed to the new disease category termed ciliopathies (
77,
98). Primary cilia are finger-like projections from the cell, enclosed in the plasma membrane, that are involved in mechanosensation, calcium influx, hedgehog and Wnt signaling, and planar cell polarity (
99). Cilia are anchored internally by a modified centriole, termed the basal body, and it is likely that the whole cilium/basal body/centriole complex is crucial for the normal maintenance of well-differentiated cells. Defects throughout this complex cause cystic kidney diseases in human and many experimental animal models (
100), but it must be pointed out that many of cyst-related proteins also have important roles in other subcellular locations such as basolateral membranes, endoplasmic reticulum, and the Golgi apparatus; hence, despite the overwhelming rush of publications, defects in the cilia/centrosome complex may not be the sole cause of cystic kidneys. Irrespective of the underlying cause, there are several fundamental abnormalities in diseased renal epithelia that eventually generate cystic kidneys, including increased proliferation in cyst epithelia coupled with apoptosis of surrounding tissues, altered polarity of cyst epithelia with mislocalization of receptor and transporter proteins, and abnormal cell-cell/cell-matrix interactions. The net effect of these is to promote cyst growth while disrupting normal surrounding tissues and renal function. Here, we consider the genetics and biology of cystic kidneys, linking cilia, and diverse pathways to these fundamental other processes, first in relation to polycystic kidneys and ciliopathies and then in cystic dysplastic and multicystic dysplastic kidneys.
Autosomal Dominant Polycystic Kidney Disease
The
PKD1 gene is located on chromosome 16p13.3 and consists of a large 53-kb genomic DNA with 46 exons. Six pseudogenes caused by duplication of PKD1 exons 1-33 are found on 16p13.1. These hampered the initial search for the gene, but they have suboptimal start codons, so they are not translated. PKD1 encodes a 460-kDa protein with over 3,000 amino acids, polycystin 1. PKD2 maps to chromosome 4q13-q23, consists of a smaller DNA sequence of 5.4 kb with 15 exons that encodes polycystin 2, a 110-kDa protein with 968 amino acids. Only one of the
PKD1 or
2 alleles is usually mutated when assessed in the blood of PKD patients, but it has long been supposed that loss of the second normal copy within the kidney is required to generate cysts (and similarly to generate PKD pathology in other organs). This is the two-hit hypothesis, but incomplete penetrance of homozygous variants and compound heterozygotes may also occur (
101), and a potential third hit such as renal injury may accelerate PKD (
102).
Polycystin 1 has (a) a large extracellular N-terminal region with many different domains including 12-15 PKD repeat motifs (immunoglobulin-like folds), two leucine-rich repeats, a region with homology to C-type lectins, and WSC, GPS, and REJ domains, (b) multiple transmembrane domains, and (c) a small intracellular 197-amino-acid cytoplasmic tail containing a coiled-coil- and G protein-binding domain. Polycystin 1
is strongly expressed in developing renal epithelia, particularly proximal tubules, and then at lower levels in the mature kidney. Expression has also been recorded in the brain, heart, and liver. At a subcellular level, polycystin 1 localizes to the cilium, lateral cell junctions, and the basolateral membrane (
103). Immunohistochemistry can still pick up polycystin 1 protein in some ADPKD kidneys, but this tends to be in cytoplasmic pools rather than associated with the cell membrane.
Polycystin 2 contains six transmembrane domains, but both its N and C termini are intracellular. It localizes to distal tubules, collecting duct, and thick ascending limb in both developing and mature kidneys. Subcellular distribution includes cilia and some membrane overlap with polycystin 1, but it is also often found in the endoplasmic reticulum, suggesting additional possible intracellular roles.
Many different functions have been ascribed to polycystin 1 and 2, both acting together and individually. When first described, polycystin 1 was linked to cell-cell and cell-matrix adhesion, whereas polycystin 2 was predicted to be an ion channel. Interestingly, this latter function was later proven correct but it requires heterodimerization of polycystin 1 and 2 together via their coiled-coil domains for calcium-permeable nonselective cation currents. Mutation of either polycystin disrupts this channel activity, and polycystin 2 is only translocated to the plasma membrane when polycystin 1 is present in the cell (
104). Other overlapping functions for the polycystins include activation of the JAK-STAT pathway by polycystin 1 with polcystin-2 acting as a critical cofactor (
105), regulation of G protein signaling, and activation of the AP1 transcription factor (
106).
Autosomal Recessive Polycystic Kidney Disease
Mutations in the polycystic kidney and hepatic disease 1 gene (
PKHD1) cause ARPKD; this is located on chromosome 6p21, spanning 470 kb and produces a 16-kb transcript. The resulting protein, termed fibrocystin or polyductin by different authors, contains 4074 amino acids with a large N-terminal extracellular region, a single transmembrane section, and a short intracellular cytoplasmic domain. In the kidney, it is mainly localized on the primary cilium and in basal bodies and the plasma membrane, in renal epithelia. Expression has also been noted in cholangiocytes. Aside from potential roles in cilia, fibrocystin may have receptor-like function via the large extracellular domain, while the intracellular part can be cleaved and translocated to the nucleus in cells stimulated with protein kinase C or increased intracellular calcium (again linking back to cilia). Several authors have reported colocalization of fibrocystin with polycystin 2, which suggests the possibility of a large PKD complex involved in a common molecular pathway in vivo (
107,
108).
Large number of mutations spread throughout the
PKHD1 gene, and the majority of patients are compound heterozygotes. There is a high risk of fetal presentation and neonatal death if the child carries two truncating mutations (
109). Cysts only derive from the collecting ducts in ARPKD but can come from all nephron segments in ADPKD (although 85% to 90% still arises from collecting ducts!). ARPKD typically presents antenatally because of ultrasonically bright, enlarged kidneys. There is a high chance of perinatal death from immature lung development if there is severe oligo- or anhydramnios before 24 weeks of gestation (
110).
Other Ciliopathies with Renal Cysts
Pathways disrupted in various NPHP include canonical and noncanonical Wnt signaling, Sonic hedgehog, and Hippo (
80,
81). Inversin, the gene product of NPHP1, illustrates the complexity and interactions within this group: inversin localizes with Nephrocystin and β-tubulin in primary cilia (
82) and acts as a molecular switch between different Wnt signaling cascades by inhibiting the canonical Wnt pathway via increased turnover of disheveled, which is downstream of the frizzled Wnt receptor, while promoting noncanonical signaling required for convergent extension (at least in
Xenopus) (
83).
 Development of the Kidney
Development of the Kidney
 Focal Segmental Glomerulosclerosis
Focal Segmental Glomerulosclerosis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis



