C3 Glomerulopathies, Including Dense Deposit Disease
H. Terence Cook
Matthew C. Pickering
C3 glomerulopathy is a recently introduced term (1) that encompasses glomerular disease characterized by the accumulation in the glomeruli of C3 or its metabolites without significant deposition of the early components of the classical pathway of complement activation, C1q and C4, and with minimal or no immunoglobulin. On electron microscopy (EM), there are electron-dense deposits corresponding to the C3 deposits seen on immunohistochemistry. The deposition of C3 with little or no immunoglobulin and without classical pathway complement activation imply uncontrolled activation of the alternative pathway of complement. Recent work has elucidated the mechanism by which this occurs in many cases. C3 glomerulopathy, thus defined, is distinct from forms of thrombotic microangiopathy that may also be associated with alternative pathway activation because in those cases complement activation is on the renal endothelium and is not associated with well-defined deposits on EM. C3 glomerulopathy also is distinct from glomerulonephritis caused by immune complex accumulation in glomeruli that contain immunoglobulin as well as complement, often including classical pathway components such as C4 or C1q. As described in more detail in the following sections, the morphology seen by light microscopy is variable and includes mesangial proliferation, a membranoproliferative pattern, endocapillary proliferation, and crescentic glomerulonephritis.
It is possible to subclassify C3 glomerulopathy based on differences of morphology as seen by light microscopy and EM (Table 9.1) or by etiology; for example, some cases are caused by an underlying gene mutation and others by autoimmune processes. Perhaps, the simplest distinction in terms of morphology is on the basis of the appearance of the electron-dense deposits seen on EM. In some cases, these have a very dense osmiophilic appearance (Fig. 9.1) leading to the designation as dense deposit disease (DDD), whereas in other cases, the deposits do not show this appearance and are designated C3 glomerulonephritis (C3GN). We therefore discuss C3 glomerulopathy in terms of these two categories while recognizing that the distinction is not always clear cut and that there is overlap of pathogenesis.
DENSE DEPOSIT DISEASE
In 1963, Berger and Galle (2) described a type of glomerulonephritis with unique, extremely osmiophilic electron-dense deposits in the glomerular basement membranes (GBMs) as the major identifying characteristic (see Fig. 9.1). Since that time, many additional reports of DDD have appeared (3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18). Because DDD often has light microscopic features of a membranoproliferative glomerulonephritis (MPGN), it has also been referred to as MPGN type II. However, it is clear that many cases do not have an MPGN pattern on light microscopy, and therefore the name DDD is to be preferred. In some series, it has accounted for up to a third of cases of MPGN although the frequency compared to type I MPGN in most series is much lower. Appel et al. (19) published an excellent review on DDD in 2005.
Clinical Presentation
DDD has a prevalence of 2 to 3 per million population and is primarily a disease of children and young adults (11,15,17,19). However, in a 2009 series from New York, 39% of the adult patients were over 60 years of age (16). It affects males and females equally in many cohorts although some studies have shown a female predominance (15,16). The clinical picture is not distinctive, and it is not possible to distinguish DDD from
immune complex MPGN type I on clinical grounds alone (5,7,10,19,20).
immune complex MPGN type I on clinical grounds alone (5,7,10,19,20).
TABLE 9.1 Pathologic classification of C3 glomerulopathy | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
At presentation, almost all patients have proteinuria (6,16,18) usually with hematuria (16,17,18). Nephrotic-range proteinuria is present in two thirds of the patients (15,16), and full nephrotic syndrome in 12% to 65% in different series (5,8,15,16,17,21). In a large series of 98 patients from North America (18), the commonest symptoms leading to health care referral were hematuria (42.9%) and peripheral (37.8%) and facial (31.6%) edema. About one fifth of these patients (21.4%) did not suspect a problem, and in this group, signs of kidney disease were detected as part of a routine annual examination. A number of patients have initial signs and symptoms of acute nephritic syndrome; this presentation is found in 16% (21) to 38% (7) of patients. There may be episodes of acute nephritis (gross hematuria, edema, hypertension) or renal insufficiency that are reversible and show complete clinical subsidence (7). Renal insufficiency is common at presentation (6,16) and is more common in adults (16). Hypertension is commonly found either at clinical onset or during the course of the disease (18).
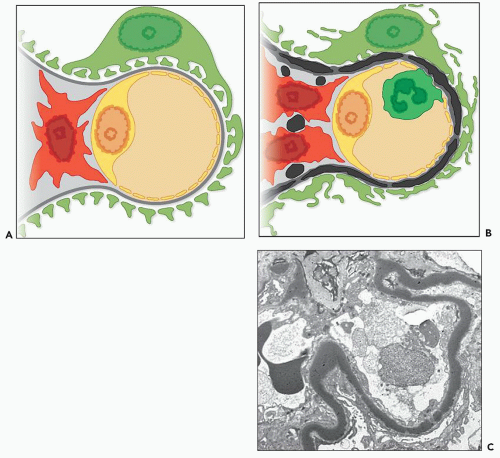 FIGURE 9.1 Diagram depicting one normal glomerular capillary (A) and a diagram (B) and electron micrograph (C) of a glomerular capillary with DDD with extensive dense deposits within the GBM as well as dense deposits in the mesangial matrix. (Green, podocyte; dark gray, GBM; yellow, endothelial cell; red, mesangial cell; light gray, mesangial matrix.) |
The clinical onset of DDD is preceded by acute infection, often an upper respiratory tract infection, in approximately one half of the patients (5,6,7,16). Elevated antistreptolysin O (ASO) titers were noted in 21% to 45% of patients with infective episodes (5,6,21); therefore, some were perhaps related to group A streptococcal infection.
Persistently, low serum levels of C3 are found in most patients (approximately 80%) (6,22), whereas fluctuating levels are found in others. In the series reported by Nasr et al. (16), the entire pediatric group had low C3 levels but significantly fewer of the adults (41%). In contrast to MPGN type I, serum levels of the early components of the classic pathway (C1q and C4) are usually normal. These complement profiles suggest that the complement is activated in DDD primarily through the alternative pathway, whereas MPGN type I arises through the classic and alternative pathways. Most patients with DDD are positive for serum C3 nephritic factor (C3NeF), an autoantibody directed against C3bBb, the convertase of the alternative pathway of complement activation. In more than 50% of patients, serum C3NeF persists throughout the clinical course (23). However, C3NeF is not a specific serologic marker because it also occurs with MPGN type I, lupus nephritis, and poststreptococcal glomerulonephritis, although less frequently. Its role is discussed further in the section dealing with pathogenesis.
Patients with DDD may also develop ocular drusen (24,25,26). These are lipoproteinaceous deposits of complement-containing debris within the Bruch membrane beneath the retinal pigment epithelium. This pathology is similar to age-related macular degeneration (AMD), but in contrast to AMD, drusen in DDD occur at an early age and may be found in the second decade of life. The long-term risk for visual problems is 10%. There is no correlation between the severity of the disease in the kidney and that in the eye.
A small minority of patients with DDD have acquired partial lipodystrophy (APL), a condition with symmetrical loss of adipose tissue from the face, arms, and upper portions of the trunk (27,28,29,30). APL is often accompanied by low levels of serum C3, normal serum levels of the early components of the complement, and the presence of C3NeF in the serum (27,28,31,32,33). Misra et al. (27) reported that approximately 83% of APL patients have low C3 levels and polyclonal C3NeF and that approximately 20% go on to develop MPGN.
Recently, an intriguing familial association has been shown between DDD and type 1 diabetes mellitus. In a series of 98 patients from North America, 16% reported at least one family member with type 1 diabetes, which is far greater than expected on the basis of the prevalence of type 1 diabetes in the general population (18). The significance of this observation remains to be determined.
Pathologic Findings
Light Microscopy
GLOMERULI
DDD is defined by the presence of dense osmiophilic transformation of the GBM on EM (see Fig. 9.1), and on light microscopy, the morphology is variable (see Table 9.1). Previous reports have emphasized the membranoproliferative form of the disease, designating this as MPGN type II, and this makes it difficult in older reports to assess the full range of histologic appearances. While it is clear that a membranoproliferative pattern of glomerular injury with increased lobulation, mesangial expansion, and capillary wall thickening with segmental double contours is common, a range of other patterns of glomerular involvement also occur (Figs. 9.2, 9.3, 9.4, 9.5, 9.6, 9.7). At one end of the spectrum, the glomeruli may show only mesangial expansion and hypercellularity. In some cases, the glomeruli show prominent endocapillary hypercellularity with segmental neutrophil infiltration (sometimes called exudative pattern). In others, large numbers of crescents may be present, warranting the diagnosis of crescentic glomerulonephritis. Walker et al. (34) collected 69 cases of DDD from centers in North America, Europe, and Japan. They identified four distinct histologic patterns on light microscopy: membranoproliferative (25%), mesangial proliferative (45%), crescentic (18%), and acute proliferative and exudative (12%). Crescentic GN was defined as more than 50% crescents. Two of their cases could not be further classified. In the Columbia series of 32 cases (16), the frequencies were MPGN (44%), mesangial proliferative (28%), endocapillary proliferative (19%), and crescentic
GN (9%). These reports emphasize that fewer than 50% of cases of DDD have an MPGN morphology. Focal crescents are common in patients without full-blown crescentic GN (16,34).
GN (9%). These reports emphasize that fewer than 50% of cases of DDD have an MPGN morphology. Focal crescents are common in patients without full-blown crescentic GN (16,34).
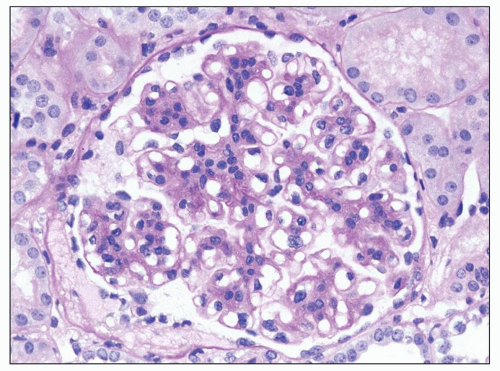 FIGURE 9.2 Glomerulus from a patient with DDD showing mesangial hypercellularity. (PAS.) (Courtesy of Dr. Patrick Walker.) |
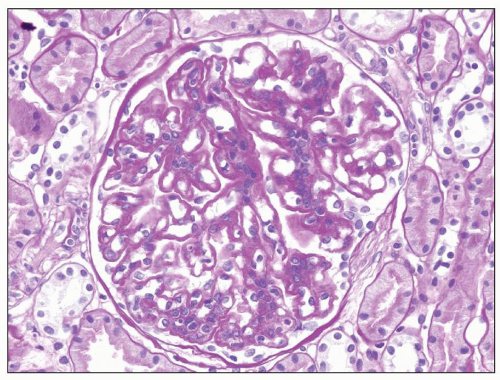 FIGURE 9.3 Glomerulus from a patient with DDD showing mesangial hypercellularity and a ribbon-like thickening of the glomerular capillary wall corresponding to the deposits seen by EM. (PAS.) (Courtesy of Dr. Patrick Walker.) |
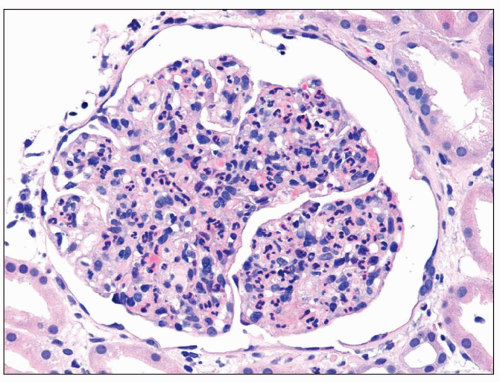 FIGURE 9.4 Glomerulus from a patient with DDD showing an endocapillary proliferative pattern with many neutrophils within capillary lumens. (H&E.) (Courtesy of Dr. Patrick Walker.) |
The GBM dense deposits are recognized on light microscopy as thickening of the GBMs by ribbon-like glassy intramembranous deposits. They stain strongly with eosin and appear somewhat refractile (hyaline). They are intensely periodic acid-Schiff (PAS) positive (see Fig. 9.3), and the trichrome stain shows them to be fuchsinophilic (red) although this reactivity varies among specimens. These glomerular intramembranous deposits appear dark blue with toluidine blue in 0.5- or 1.0-µm plastic-embedded sections. With methenamine silver staining, the intramembranous deposits are not as argyrophilic as the GBM and are somewhat brownish (6,35). Thus, the intramembranous nonstaining deposit is often bordered on each side by a thin, silver-positive GBM (36). A double-contour appearance of the glomerular capillary wall is noted in approximately half of the patients. The glomerular intramembranous deposits may stain with the fluorochrome dye thioflavin T (Fig. 9.8) (9,37).
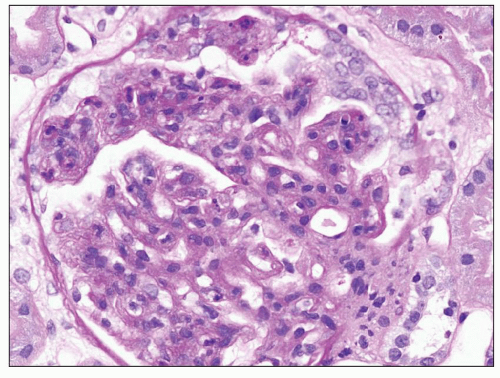 FIGURE 9.5 Glomerulus from a patient with DDD showing prominent segmental hypercellularity with endocapillary hypercellularity and a small cellular crescent. Some capillary walls show a double-contour appearance. (PAS.) (Courtesy of Dr. Patrick Walker.) |
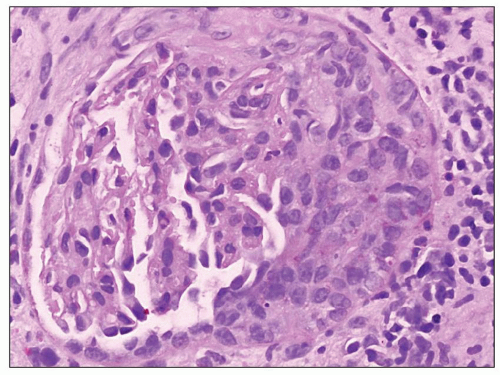 FIGURE 9.6 Glomerulus from a patient with DDD showing a cellular crescent. (PAS.) (Courtesy of Dr. Patrick Walker.) |
The glomerular intramembranous deposits vary greatly in size and number, and it is this feature that determines the extent of the glomerular capillary wall thickening. In typical cases, diagnosis may be suspected by light microscopy although EM may be needed to establish the presence of diagnostic deposits with certainty. In some biopsies, PASand trichrome-positive mesangial deposits are evident. EM often reveals mesangial deposits that cannot be identified by light microscopy. Deposits are also noted in the basal lamina of the Bowman capsule and the tubular basement membranes
(TBMs) of both proximal and distal tubules (Fig. 9.9) (38); the extent of this extraglomerular involvement is quite variable.
(TBMs) of both proximal and distal tubules (Fig. 9.9) (38); the extent of this extraglomerular involvement is quite variable.
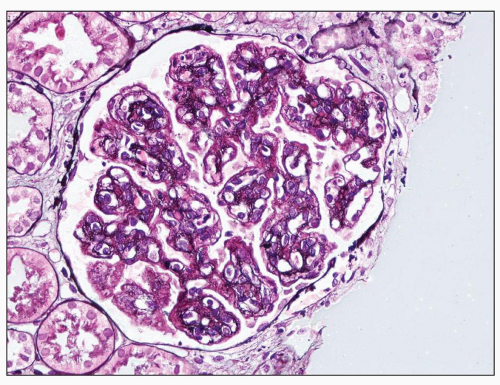 FIGURE 9.7 Glomerulus from a patient with DDD showing MPGN patterns. There are mesangial increase and capillary wall double contours. (Jones methenamine silver.) |
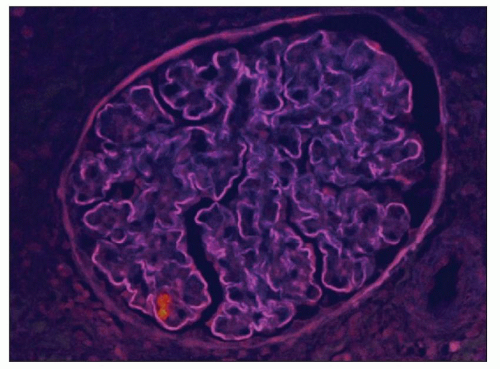 FIGURE 9.8 Glomerulus from a patient with DDD showing the staining of the capillary wall dense deposits with thioflavin T. (Thioflavin T stain.) (Courtesy of Dr. Patrick Walker.) |
As renal disease progresses, the glomeruli undergo sclerosis, but even in advanced cases, it is usually possible to see the characteristic intramembranous dense deposits by EM. In the Columbia series, segmental glomerulosclerosis was significantly more common with the MPGN pattern of GN and also in adults compared with children (16).
TUBULES
The tubular epithelium may display either hyaline droplets (evidence of glomerular filtration of protein) or vacuolation. The same type of deposit noted in the GBM may be identified along the proximal and distal TBMs (38) (see Fig. 9.9). With progression of glomerular sclerosis, there are tubular atrophy and interstitial fibrosis.
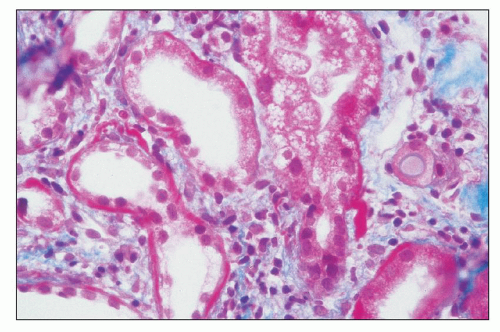 FIGURE 9.9 Focal band-like fuchsinophilic (red) deposits in the tubular basement membranes corresponding to the dense deposits seen by EM. (Trichrome, ×400.) |
BLOOD VESSELS
INTERSTITIUM
Foam cells are sometimes present, indicative of the nephrotic syndrome with substantial hypercholesterolemia and lipiduria. Interstitial fibrosis appears as renal failure develops.
Immunohistochemistry
The invariable finding in DDD is the presence of C3 in the glomeruli (5,6,16,34,39,40,41). Intense staining for C3 is noted along the glomerular capillary walls and often in the glomerular mesangial regions (Fig. 9.10). The immunofluorescence patterns have been variously described as linear, pseudolinear, smooth, ribbon-like, granular, or nodular. The C3 deposition is usually diffuse and global. The GBM staining may be continuous or discontinuous. The mesangial deposits may stain as scattered granules, coarse granules, spherules, or small rings. The early components of complement, C1q and C4, are usually absent, although occasionally C1q is found (16,34,42). One study (43) reported C4 in 9 of 13 patients when glomerular granular capillary wall staining was present. When there is staining for C1q or C4, it is less intense than C3 and may be focal and segmental (38). C3 may also be detected in the Bowman capsule and along the TBMs.
Kim et al. (40) have reported a characteristic staining pattern for C3. A double-linear appearance, referred to as “railroad tracks,” was detected in the glomerular capillary walls, while numerous circular, unevenly stained structures termed mesangial rings were noted in the mesangial areas. Using phase-contrast and electron microscopy with a double-labeling antibody technique (rhodamine-conjugated anti-GBM and fluoresceinated anti-C3), these authors thought that the railroad tracks and the mesangial rings represented the deposition of C3 along the
margin, but not within the central portions, of the large intramembranous or mesangial deposits (40). However, our own unpublished data using immunoelectron microscopy showed staining of the dense deposit itself for C3 and C9. It may be that the pattern depends on the fragment(s) of C3 detected by the antibodies, and this is worthy of further study.
margin, but not within the central portions, of the large intramembranous or mesangial deposits (40). However, our own unpublished data using immunoelectron microscopy showed staining of the dense deposit itself for C3 and C9. It may be that the pattern depends on the fragment(s) of C3 detected by the antibodies, and this is worthy of further study.
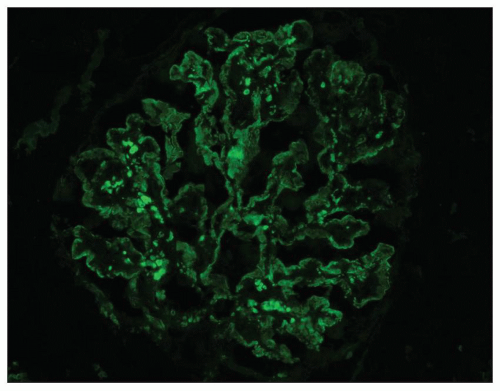 FIGURE 9.10 Immunofluorescence with antibody to C3c in a patient with dense deposits disease. There is staining of the glomerular capillary wall and also staining in the mesangium of spherical structures some of which have accentuation of staining at the edges (ring pattern). |
Immunoglobulins are usually absent or show only focal and segmental staining. If they are present, they often stain much less intensely than C3 and they are usually of the IgM type with a segmental distribution; IgG and especially IgA are less common (16,40). Joh et al. (38) suggested that IgG deposition in DDD is related to the glomerular subepithelial deposits visible on EM. Using a monoclonal antibody to a neoantigen of the C9 portion of the membrane attack complex (MAC) of complement, Falk et al. (44) performed immunofluorescence and immunoelectron microscopy to study the antigen distribution in renal biopsies of DDD. MAC and C3 were distributed along either side of the dense deposits within the GBMs and TBMs, around the circular mesangial deposits, and within the subepithelial deposits. In 12 biopsies from patients with DDD and an MPGN pattern, properdin staining was seen in 3 and was absent in 9 specimens (41), whereas Kim et al. (40) found properdin in a granular distribution in all 9 biopsies they examined and along the capillary wall in 5.
Electron Microscopy
EM led to the discovery of DDD, which is distinct from other forms of renal disease (2). The disease takes the name “dense deposit disease” from the characteristic ultrastructural appearance of the glomerular intramembranous deposits (Figs. 9.1 and 9.11). There have been many reports on the electron microscopic findings (4,5,6,9,10,11,16,34,36,38), and there is generally good agreement on the ultrastructural features. Confirmation of the diagnosis of DDD requires EM, although the diagnosis can be suspected with high confidence if the typical light microscopic and immunofluorescence microscopic features are observed.
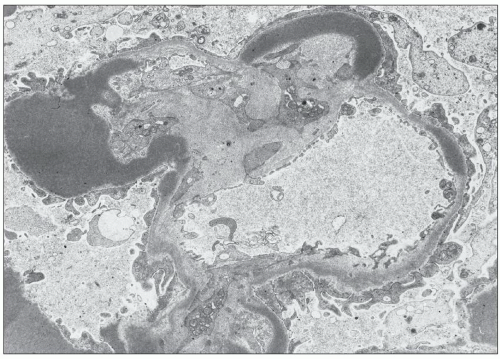 FIGURE 9.11 Dense deposits in the GBM. They are of a much greater density than the lamina densa and are usually elongated, as in this photo. (×3200.) (Courtesy of Dr T. Antonovych.)
Related posts: Development of the Kidney Development of the Kidney
 Membranous Glomerulonephritis Membranous Glomerulonephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
 Renal Disease Caused by Hypertension Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|