Alport Syndrome, Familial Benign Hematuria, Nail-Patella Syndrome, Type III Collagen Glomerulopathy, and Pierson Syndrome
Marie Claire Gubler
Laurence Heidet
Corinne Antignac
ALPORT SYNDROME
In 1927, Alport (1) reported the association of deafness with a form of “hereditary familial congenital hemorrhagic nephritis” occurring in successive generations of one family. He observed that “the male members tend to develop nephritis and deafness and do not as a rule survive,” whereas “the females have deafness and hematuria and live to old age.” The main features of the disease were established, and an X-linked dominant inheritance, the usual mode of inheritance, was clearly suggested. Subsequently, many additional families have been reported from various parts of the world, and the clinical spectrum and genetic basis of Alport syndrome (AS) have expanded to include other extrarenal defects and variants. The estimated frequency of AS is 1:5000. It accounts for 1% to 2% of end-stage renal disease (ESRD) in Europe, India, and the United States.
The pathogenesis of AS has been elucidated by advances in pathology, biochemistry, genetics, and molecular biology. The primary defect in AS involves type IV collagen, the main component of the basement membranes. AS is genetically heterogeneous, and different mutations in the genes encoding the α3, α4, or α5 chain of type IV collagen are responsible for the various forms of disease. X-linked inheritance is the most common mode of transmission. The phenotypic heterogeneity of the disease can be explained by the molecular heterogeneity: nearly every family has its “private” mutation. Benign familial hematuria may represent a benign variant of type IV
collagen diseases and the heterozygous form of autosomal recessive AS. Animal models of AS now provide opportunities for investigating pathogenesis of the disease and developing new therapeutic approaches.
collagen diseases and the heterozygous form of autosomal recessive AS. Animal models of AS now provide opportunities for investigating pathogenesis of the disease and developing new therapeutic approaches.
Pathologic Findings
Gross Appearance
No specific macroscopic changes have been reported in the kidneys obtained at autopsy of patients dying from ESRD. The kidneys are shrunken and atrophic with a finely granular cortical surface (2). Atrophy predominates in the cortex, in which yellow linear streaks are sometimes visible (corresponding to lipid-filled foam cells). More severe contraction is observed in the kidneys obtained after several years of dialysis.
Light Microscopy
GLOMERULI
In renal biopsies obtained early in the course of the disease, the glomeruli appear normal or show minimal changes, hypertrophy of podocytes, rigidity of glomerular capillary walls, irregular enlargement of mesangial stalks, and focal thickening of the capsular basement membrane (Fig. 13.1A and B). Focal and segmental thickening and lamellation of the glomerular capillary walls, better seen on silver stains, become visible with progression of the disease, and they are usually associated with marked widening of the mesangial stalks. Segmental lesions of the tuft, producing a pattern of focal and segmental glomerulosclerosis (FSGS), develop in an increasing number of glomeruli; they are caused by marked thickening of the glomerular basement membrane (GBM) and the mesangial stalks, associated with hyaline deposits and collapse of the capillary loops (Fig. 13.1C and D). The end point is complete sclerosis or hyalinosis of the glomerulus. Correlation between the severity of glomerular changes and the age at renal biopsy has been observed by several groups (4,6).
TUBULES
The tubules are initially normal, except for the presence of occasional red blood cell casts (Fig. 13.1E). The foci of nonspecific tubular lesions then develop. These lesions actually may precede significant light microscopic glomerular changes but subsequently develop alongside the glomerular lesions.
INTERSTITIUM
Interstitial fibrosis is usually absent before the age of 10 years. Then, interstitial fibrosis and tubular atrophy develop and progress in correlation with declining glomerular filtration rate (GFR) (3). Focal areas of interstitial fibrosis may be seen before the appearance of overt glomerular changes.
Lipid-laden foam cells have been considered a marker of AS because of their abundance and distribution (Fig. 13.1F). When present, they are concentrated at the corticomedullary junction or they occur as long, linear rows throughout the cortex. However, they are inconstant and are rarely found in early biopsy specimens. They are abundant at the stage of heavy proteinuria, and their number may decrease with progression to renal failure. They are not specific and are observed in patients with abundant proteinuria, regardless of the type of glomerular involvement.
BLOOD VESSELS
No significant vascular changes are observed early in the course of the disease. Lesions of arteriolosclerosis may develop in hypertensive patients.
Immunofluorescence Microscopy
Immunofluorescence microscopy of renal tissue using antibodies to immunoglobulins and complement components is usually negative (6,7). However, faint deposits of IgG, IgM, and the third component of complement (C3) are occasionally observed (3,4,5,7) and may mimic immune complex-mediated glomerulonephritis in a few patients (8). Scattered granules of C3 distributed haphazardly throughout the glomerular tuft and the arterioles are not uncommon, but their presence is not a marker of any specific disease and their absence does not exclude the diagnosis of AS. With progression of the disease, focal deposits of IgM, C3, or C1q are observed in hyalinized segments of glomeruli.
Electron Microscopy
Ultrastructural studies demonstrated the basic lesion of AS (5,9,10,11), subsequently observed by many investigators (3,4,6,7,12,13). This finding was of the utmost importance because it provided a useful morphologic marker for AS and gave the first indication of the primary involvement of the GBM in the disease.
The typical lesion is characterized by thickening of the GBM (800 to 1200 nm), with splitting and fragmenting of the lamina densa into several strands forming a basket-weave pattern. Small, electron-dense granules are usually visible within the lucent zones delineated by the thin interwoven layers of lamina densa (Figs. 13.2 and 13.3). The inner and outer contours are irregularly festooned and are lined by hypertrophied podocytes on the exterior. The lesion is often widespread, at least in adults, involving more than 50% of capillary loops. It seems to become more extensive on serial biopsy specimens (5,14). However, GBM changes may be patchy, alternating with segments of normal or reduced thickness. In fact, in younger patients, a second type of lesion, the thin basement membrane (100 to 200 nm), is the prevalent change (4,6,7,12,13,14), and focal ruptures of the GBM with repair by newly formed basement membrane material may be seen (4). On the whole, the most striking feature in children is the irregular appearance of the GBM, resulting from the close alternation, even within the same loop, of thick, split, and extremely thin GBM segments (Fig. 13.4). The GBM lesion develops early in life; it has been found in several patients younger than 2 years. It is observed in male and female patients, but as described by Rumpelt (14) in a quantitative ultrastructural analysis, and confirmed by White et al. (13), male patients have more splitting than do female patients, and splitting increases with age in male patients but not in female patients. Similar findings have been reported in the Samoyed dog, an animal model of X-linked AS: The GBM is normal in thickness at birth in both sexes; then, extensive thickening and splitting of the GBM appear in the males, whereas only focal areas of multilaminar splitting occur in females (15).
In some patients, the typical GBM thickening is absent (9,12) and widespread thinning of the GBM is observed, whatever the age of the patient and the type of the mutation (Figs. 13.5 and 13.6) (6,7,12,16,17,18). In a series of 105 patients
affected with progressive hereditary nephritis and studied by electron microscopy, we observed diffuse attenuation of the GBM in 30; 19 of these patients had classic AS, and 11 had progressive hereditary nephritis without deafness. Intrafamilial concordance of ultrastructural changes is usually observed, and thickening of the GBM with fragmentation of the lamina densa is both a marker of certain families and an index of severity. However, rare discordances have been reported (12,19).
affected with progressive hereditary nephritis and studied by electron microscopy, we observed diffuse attenuation of the GBM in 30; 19 of these patients had classic AS, and 11 had progressive hereditary nephritis without deafness. Intrafamilial concordance of ultrastructural changes is usually observed, and thickening of the GBM with fragmentation of the lamina densa is both a marker of certain families and an index of severity. However, rare discordances have been reported (12,19).
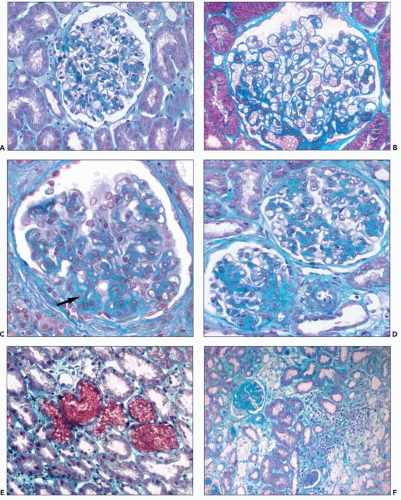 FIGURE 13.1 Alport syndrome. Light microscopy. A: Normal glomerulus in a 6-year-old boy. (×250.) B: Rigidity and moderate thickening of the GBM in a 12-year-old boy. (×330.) C: Irregular thickening of the basement membrane, marked podocyte hypertrophy, and focal segmental glomerulosclerosis (arrow) in a 14-year-old boy. (×330.) D: Multiple lesions of focal segmental glomerulosclerosis in a 17-year-old boy. (×250.) E: Presence of red blood cell casts. (×350.) F: Clusters of interstitial foam cells. Same patient as in (D). (×200.). (A-F, trichrome light green.) |
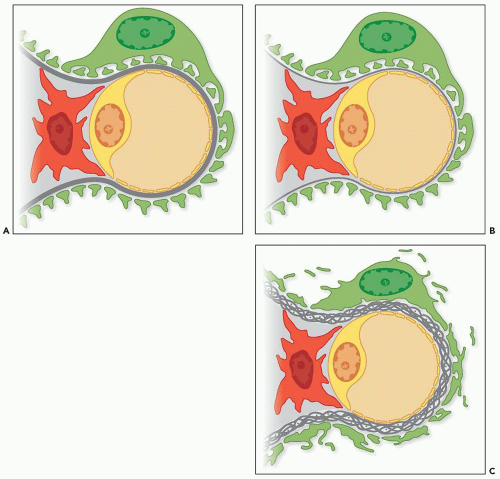 FIGURE 13.2 Diagram depicting one normal glomerular capillary (A), a capillary with a thin basement membrane lesion that can be seen in Alport syndrome and in thin basement membrane nephropathy (B), and a capillary with irregular GBM lamination representing the changes of Alport syndrome (C) (green, podocyte; dark gray, GBM; yellow, endothelial cell; red, mesangial cell; light gray, mesangial matrix). |
Other lesions have been described. Frequently, podocytes are enlarged, contain vacuoles, and show focal or diffuse effacement of foot processes. The mesangium is initially normal, followed by progressive increase in mesangial matrix, focal mesangial hypercellularity, and mesangial expansion along the subendothelial aspect of the GBM. The mesangial material appears heterogeneous, containing dense granules of various sizes, clear areas, and curvilinear membranous structures. Finely granular dense deposits can be found focally in severely involved GBM segments (Fig. 13.7). Focal thickening of the Bowman capsule or tubular basement membrane (TBM) is frequently associated with the GBM lesions (Fig. 13.8). These thickenings are characterized by marked bulging and splitting of the basement membranes delineating clear areas containing vesicular structures or dense laminated particles having the appearance of lipid deposits. Accumulation of lipid droplets within capsular epithelial cells is rarely observed, whereas it is frequent in tubular and interstitial cells.
The specificity of the GBM pathologic features has been questioned. According to Hill et al. (20), thickening and splitting of the GBM closely mimicking hereditary nephritis can be seen in poststreptococcal glomerulonephritis, FSGS and IgA nephropathy, and GBM attenuation may be observed in most types of glomerulopathies. Actually, widespread
thickening and splitting of the GBM in the proper clinical setting strongly support a diagnosis of AS, but occasional areas of splitting clearly do not have the same diagnostic specificity. In our experience, the finding of extensive GBM changes in 30 of 50 hematuric children, in whom the diagnosis of IgA nephritis had been ruled out by immunofluorescence, allowed the diagnosis of AS, which was confirmed by further family investigations, follow-up data, or the finding of type IV collagen mutations. Characteristic ultrastructural changes of the GBM have been observed in patients with no family history of renal or hearing impairment (5). These observations are important because they strongly suggest that these patients are affected with de novo AS, a hypothesis easily confirmed by genetic studies. Very rare cases of widespread GBM lesions mimicking AS pathology have been described in young children presenting with isolated sporadic or familial steroid-resistant nephrotic syndrome (21), in a few cases of Denys-Drash or Frasier syndromes linked to WT1 mutation (22), in the rare Pierson syndrome linked to mutations in the LAMB2 gene (see section on Pierson syndrome), or in nephrotic syndrome associated with hypomelanosis of Ito (23).
thickening and splitting of the GBM in the proper clinical setting strongly support a diagnosis of AS, but occasional areas of splitting clearly do not have the same diagnostic specificity. In our experience, the finding of extensive GBM changes in 30 of 50 hematuric children, in whom the diagnosis of IgA nephritis had been ruled out by immunofluorescence, allowed the diagnosis of AS, which was confirmed by further family investigations, follow-up data, or the finding of type IV collagen mutations. Characteristic ultrastructural changes of the GBM have been observed in patients with no family history of renal or hearing impairment (5). These observations are important because they strongly suggest that these patients are affected with de novo AS, a hypothesis easily confirmed by genetic studies. Very rare cases of widespread GBM lesions mimicking AS pathology have been described in young children presenting with isolated sporadic or familial steroid-resistant nephrotic syndrome (21), in a few cases of Denys-Drash or Frasier syndromes linked to WT1 mutation (22), in the rare Pierson syndrome linked to mutations in the LAMB2 gene (see section on Pierson syndrome), or in nephrotic syndrome associated with hypomelanosis of Ito (23).
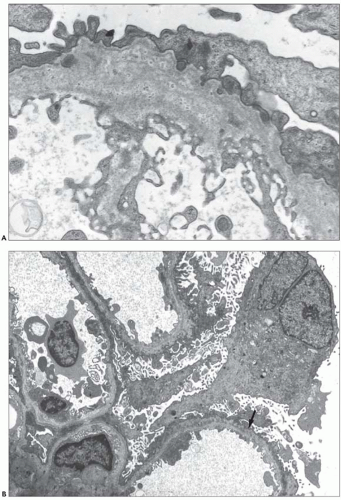 FIGURE 13.3 A: Electron microscopy from a 12-year-old boy with X-linked Alport syndrome. Note thickening and irregular contours of the glomerular basement membrane, splitting of the lamina densa, and small electron-dense granules. (Uranyl acetate-lead citrate, ×22,500.) B: Electron microscopy from the same 12-year-old boy showing the irregular distribution of GBM lesions with nearly diffuse foot process effacement and focal presence of granular deposits (arrow) distributed along a thick GBM segment. (Uranyl acetate-lead citrate, ×7800.) |
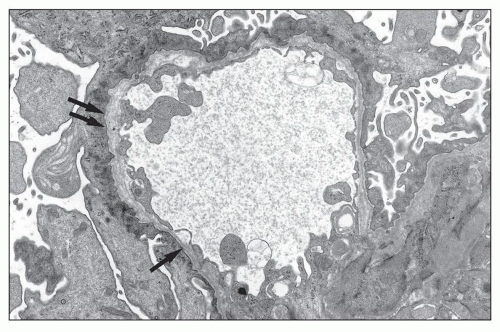 FIGURE 13.4 Electron microscopy from a 10-year-old boy with X-linked Alport syndrome. Note the alternation of thick and split (double arrows) and of thin (single arrow) glomerular basement membrane segments. (Uranyl acetate-lead citrate, ×13,200.) |
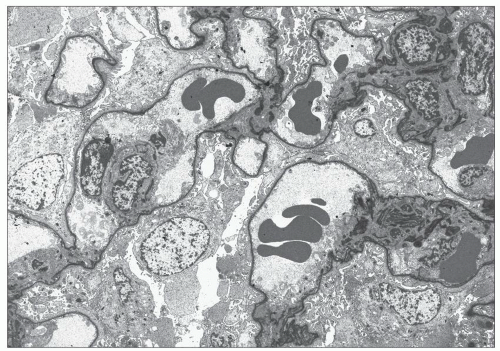 FIGURE 13.5 Electron microscopy from a 26-year-old male patient with X-linked Alport syndrome who developed ESRD at the age of 37 years. Note the widespread attenuation of the GBM. (Methenamine silver staining ×2500.) |
Extrarenal Tissues
Only a few studies have been devoted to nonrenal tissues. Unique pathologic changes have been demonstrated by postmortem light and electron microscopic examination of the inner ear. These include a zone of separation between the basilar membrane and the basement membrane of cells of the organ of Corti, and the presence of cells filling the tunnel of
Corti, morphologically similar to supporting cells (24). In the eye, the anterior capsule of the lens is thinned, and vertically oriented fractures have been seen (25,26). The retina may have thinning of the internal limiting membrane/nerve fiber layers and of the retinal pigment epithelial basement membrane of the Bruch membrane (27). Contrary to initial observations, electron microscopic examination of the epidermal basement membrane (EBM) failed to disclose any significant ultrastructural abnormalities (28,29, personal observations).
Corti, morphologically similar to supporting cells (24). In the eye, the anterior capsule of the lens is thinned, and vertically oriented fractures have been seen (25,26). The retina may have thinning of the internal limiting membrane/nerve fiber layers and of the retinal pigment epithelial basement membrane of the Bruch membrane (27). Contrary to initial observations, electron microscopic examination of the epidermal basement membrane (EBM) failed to disclose any significant ultrastructural abnormalities (28,29, personal observations).
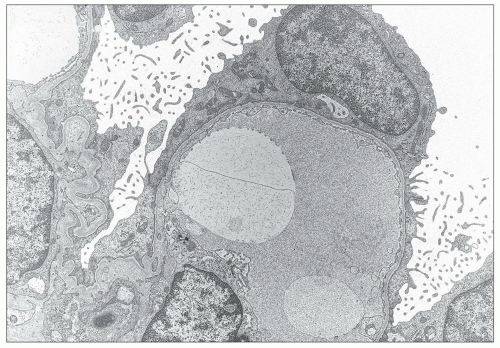 FIGURE 13.6 Electron microscopy from the same 26-year-old patient. Extremely thin GBM with regular contours and diffuse effacement of foot processes. (Uranyl acetate-lead citrate, ×8000.) |
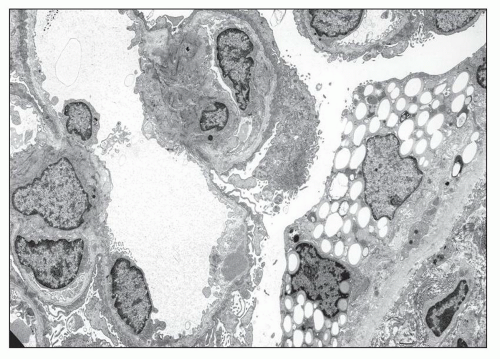 FIGURE 13.7 Electron microscopy from a 12-year-old boy with Alport syndrome showing the widespread alteration of the GBM with focal subendothelial and subepithelial deposits. There are diffuse effacement of foot processes, Bowman capsular thickening, and a foamy appearance of the capsular epithelial cell. (Uranyl acetate-lead citrate, ×4500.) |
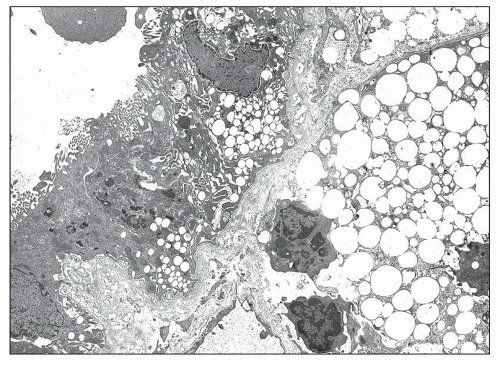 FIGURE 13.8 Electron microscopy from a 15-year-old boy with Alport syndrome showing marked TBM thickening and splitting. Lipid droplets are present in tubular epithelial cells. They massively involve the interstitial cell cytoplasm. (Uranyl acetate-lead citrate, ×4500.) |
Type IV Collagen
Type IV collagen is the major structural component of all basement membranes. Like other members of the collagen family, type IV collagen is a multimeric protein composed of three α chains coiled around one another to form a triple-helical molecule or protomer (30). Each chain comprises a long 350-nm collagenous domain and, in contrast to interstitial collagens, a noncollagenous (NC) globular domain at the 3′ end of the molecule and a short NC domain at the 5′ end. The collagenous domain, which contains about 1400 amino acid residues, is characterized by the repeated Gly-X-Y triplet sequence, in which every third amino acid is a glycine. The presence of glycine in every three residues is crucial for proper triple-helix formation, because glycine is the only amino acid small enough to fit into the center of the triple helix. Twenty-one to twenty-six short interruptions of the Gly-X-Y sequence are presumed to give flexibility to the molecule. The C-terminal NC1 domain, of about 230 amino acid residues, is a globular structure consisting of two symmetrical subdomains, each containing six cysteine residues, the positions of which are highly conserved between hemi-NC subdomains and between chains. These cysteines are the basis for intrachain and interchain disulfide bonds. Cysteine residues are also present in the short NC sequence located at the N-terminal part of the chains.
Six genetically distinct but closely related α(IV) chains have been identified (Fig. 13.9A). Each contains about 50 exons. Because of the large size of their introns, they are large genes, with a size between 100 and more than 425 kilobases (kb) (31). They are located pairwise in a head-to-head fashion on three different chromosomes. The structural similarities between the different chains suggest that the genes diverged from a common ancestor that was initially duplicated on the same chromosome and then eventually triplicated. COL4A1 and COL4A2 were the first to be localized (13q34). They are separated by 130 base pairs (bp) and share a bidirectional promoter (32). The genes COL4A3 and COL4A4 were assigned to 2q35-37, and their genetic structure was determined (33,34,35). The genes encoding the α5 and α6 chains of type IV collagen were localized to chromosome Xq22 and characterized. They are separated by a short 450-bp intergenic region (36,37,38,39).
Many type IV molecules could be potentially formed. However, only three protomers have been discovered: α1.α1. α2(IV), α3.α4.α5(IV), and α5.α5.α6(IV) (Fig. 13.9B) (31). They associate to form interwoven networks providing a scaffold for other basement membrane glycoproteins: two protomers bind via their NC1 domains to form dimers; four molecules associate via disulfide cross-links in the 7S domain to form tetramers (Fig. 13.9C). Three distinct networks have been recognized: α1α1α2(IV)-α1.α1.α2(IV), α3.α4.α5(IV)-α3.α4.α5(IV), and α1.α1.α2(IV)-α5.α5.α6(IV). Lateral sideby-side interactions, especially numerous between α3.α4. α5(IV) protomers, make the network tighter and more resistant to proteolysis (40).
The three networks are not equally distributed within the basement membrane. The most abundant, α1.α1.α2(IV) is ubiquitous and found in all basement membranes. However, in the glomerular tuft, the strong mesangial expression of the α1(IV) and α2(IV) chains contrasts with their faint GBM distribution, limited to the subendothelial aspect of the basement membrane (Fig. 13.10). The other networks have restricted and specific distributions. In the mature kidney, the α3.α4. α5(IV) network is present within the GBM, the distal tubule basement membrane, and focally in the Bowman capsule basement membrane (7,31,41,42). However, in rat as in human GBM development, there is a switch from the exclusive α1.α1. α2(IV) network detected at the early capillary loop stage to the α3.α4.α5(IV) network present in the mature glomerulus where it is synthesized by the podocyte (43). The α3.α4.α5(IV) network is also present in the alveolar basement membrane and the specialized basement membrane of the eye and the cochlea (44,45,46,47,48). The α1.α1.α2(IV)-α5.α5.α6(IV) network is expressed in the Bowman capsule and collecting duct basement membranes, in the epidermal and smooth muscle cell basement membranes of the urinary bladder, uterus, esophagus, and fundus of the stomach, and in the vascular smooth muscle cell basement membrane of the aorta (49). The α6 chain is consistently absent from the GBM.
AS is genetically heterogeneous (50). Mutations in COL4A5 result in X-linked dominant AS, whereas mutations in COL4A3 or COL4A4 lead to autosomal recessive or dominant AS. In most patients, the resulting chain defect (absence or abnormal structure) impairs the protomer assembly and the formation of the normal collagen IV network, a consequence readily observed by immunohistologic staining of the basement membranes using specific antibodies against the different α(IV) chains. Importantly, COL4A3 or COL4A4 mutations in the heterozygous state are also associated with benign familial hematuria.
X-Linked Dominant Alport Syndrome
X-linked dominant transmission is the mode of inheritance in 80% to 85% of AS families.
Clinical Features
Classic X-linked AS is characterized by the familial occurrence of hematuric nephritis progressing to ESRD, at least in male patients, and of hearing loss, which is also progressive. Hematuria is the cardinal symptom. Macroscopic or microscopic, it is usually detected in childhood, in infancy, or even at birth (2,4,11,51). In a series of 58 patients observed in our Department of Pediatric Nephrology, the age at discovery of hematuria was less than 6 years in 74% (4). However, in some patients who do not manifest macroscopic hematuria, microscopic hematuria may be detected only in adulthood, either isolated or associated with proteinuria, hypertension, or chronic renal failure.
Evolution and prognosis differ according to sex. In male patients, microscopic hematuria is universal and usually persistent. Its presence is a necessary criterion for diagnosis. Single or recurrent episodes of macroscopic hematuria precipitated by exercise or upper respiratory tract infections are observed in about 60% of the patients younger than 15 years old, but these episodes are exceptional in adults. Proteinuria may be absent, mild, or intermittent in young patients. It increases
steadily with age, with possible development of the nephrotic syndrome (4). Proteinuria was observed in 95% of the patients belonging to the European Community cohort of 195 AS families (51). All male patients progress to ESRD at ages ranging from 8 to older than 60 years. Hypertension is usually absent before the stage of chronic renal insufficiency. According to the rate of progression to ESRD, two types of the disease have been distinguished: (a) a “juvenile” type characterized by a highly stereotypical course within a given family and by the occurrence of ESRD in men around the age of 20 years and (b) a “nonprogressive” or “adult” type in which the age at ESRD is higher, approximately 40 years, and the course much more variable. In the latter group, precise prognostication in the individual case is impossible.
steadily with age, with possible development of the nephrotic syndrome (4). Proteinuria was observed in 95% of the patients belonging to the European Community cohort of 195 AS families (51). All male patients progress to ESRD at ages ranging from 8 to older than 60 years. Hypertension is usually absent before the stage of chronic renal insufficiency. According to the rate of progression to ESRD, two types of the disease have been distinguished: (a) a “juvenile” type characterized by a highly stereotypical course within a given family and by the occurrence of ESRD in men around the age of 20 years and (b) a “nonprogressive” or “adult” type in which the age at ESRD is higher, approximately 40 years, and the course much more variable. In the latter group, precise prognostication in the individual case is impossible.
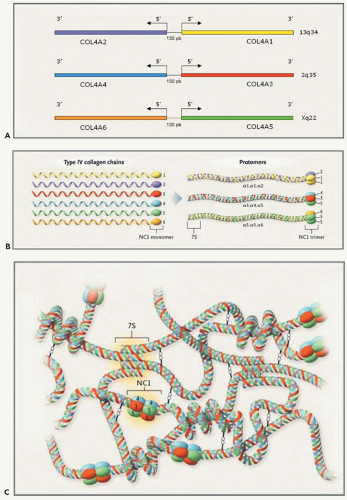 FIGURE 13.9 A: The genes COL4A1 to COL4A6 are located in pairs, head to head, on three different chromosomes. B: They encode six different type IV collagen α chains. Each of them comprises a long collagenous domain, a noncollagenous globular domain at the C terminus, and a 7S domain at the N terminus. They are arranged into three triple-helical protomers that differ in their chain composition. C: Protomers create basement membrane networks with other protomers by uniting two NC1 domains at the C terminus and by uniting four 7S domains at the N terminus. Here is shown the α3.α4.α5(IV)-α3.α4.α5(IV) network, with disulfide cross-links between triple-helical domains. (Modified from Hudson BG, Tryggvason K, Sundaramoorthy M, et al. Alport‘s syndrome, Goodpasture‘s syndrome, and type IV collagen. N Engl J Med 2003;348:2543.) |
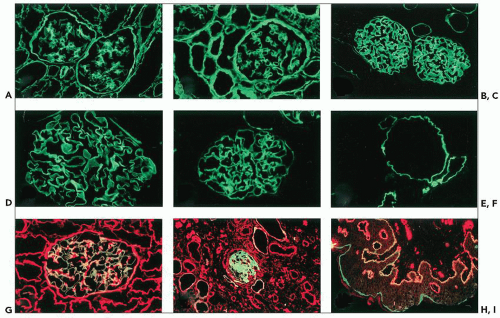 FIGURE 13.10 Immunofluorescence. A-F: Normal renal distribution of the α1-α6 type IV collagen chains. A and B: The α1 and α2 chains are codistributed in all basement membranes, but their expression is faint in the GBM. C-E: The α3, α4, and α5 chains are codistributed in the GBM and the basement membranes of distal tubules; they are absent in the mesangial matrix. F: The α6(IV) chain is absent in the GBM and strongly expressed in the Bowman capsule and the distal tubule basement membranes. G-I: Double immunolabeling with anti-α2 (Texas red) and anti-α5(IV) (fluorescein) antibodies showing the respective localization of the two chains in a normal kidney specimen (G) and the strong persistence of the α5(IV) chain in the sclerotic glomerulus from a non-AS patient (H). In the skin (I), both chains are present in the epidermal basement membrane (yellow fluorescence). |
Hematuria is observed in nearly all female patients, but it may be intermittent or detected only in adulthood. It was present in 95.5% of patients (309 of 323) in the European series (52). Completely asymptomatic carriers are rare. Proteinuria may be absent; it is usually mild or intermittent (4,5,52) and was observed in 75% of patients in the European Study (52). It increases with age in some patients at risk of progressing to ESRD. The prognosis of renal involvement is impossible to predict from family history, as shown by the clinical course of affected female members of the family initially reported by Alport: three of them developed ESRD between 12 and 24 years of age, whereas three others had a normal life span of 69 to 90 years (53). Gross hematuria in childhood, progressive increase in proteinuria, nephrotic syndrome, diffuse GBM thickening, and hearing loss are associated with an adverse outcome (54). Random X inactivation (the process known as lyonization) may account for the variable clinical course of the disease in female patients. On the whole, according to Jais et al. (52), the risk of developing ESRD before the age of 40 years was 12% in girls and women versus 90% in boys and men, but the risk of progression in women appears to increase after the age of 60 years.
Bilateral sensorineural hearing loss affecting high and middle frequencies is not congenital, but it may be detected over the first decade, especially in boys (4). In children, serial audiologic tests show progressive hearing loss in most boys and some girls, whereas hearing impairment is generally stable in adults. The true prevalence of hearing loss within AS-affected families is difficult to assess in the absence of systematic audiologic evaluation. Some kindreds with progressive hereditary nephritis without hearing loss are AS variants. They represent nearly 20% of families with COL4A5 mutations in the study of Jais et al. (51).
Several types of ocular changes involving the lens, retina, or cornea have been reported. Anterior lenticonus is a conical protrusion of the anterior aspect of the lens that develops progressively over the years in male patients (55) and is exceptional in female patients (4,53,56). Its presence is predictive of early renal failure (56). Retinal changes are characterized by the progressive appearance of asymptomatic perimacular yellowish flecks (57,58). Both types of lesions are specific and are observed in about one third of these patients. Nonspecific lesions of the cornea have been reported to occur frequently in patients with AS: posterior polymorphic dystrophy was detected in 11 of 17 patients from Thailand (59), and recurrent corneal erosions were observed in about 20% of patients with AS and renal failure (60).
Other different anomalies have been reported in patients with X-linked AS. First described by Garcia-Torres and Orozco (61), diffuse esophageal leiomyomatosis, also involving the tracheobronchial tree and the female genital tract, has now been reported in 69 patients from 33 families, all affected with the severe, juvenile type of AS. The main symptoms, dysphagia, postprandial vomiting, and recurrent episodes of bronchitis, usually appear before 10 years of age. Congenital cataracts occur in one third of these patients. Unlike AS, diffuse leiomyomatosis is fully penetrant and is completely expressed in female patients (62). Peculiar deletions of the COL4A5-COL4A6 genes have been demonstrated in these patients (63). Aortic abnormalities including dissections and aneurysms occurring between 13 and 36 years of age have been reported in eight male patients. All of them had a severe renal disease requiring renal replacement therapy between 10 and 22 years (64). The presence of the α1.α1.α2(IV)-α5.α5.α6(IV) network in the smooth muscle cell basement membrane of the digestive tract and the aorta can explain these manifestations. On the other hand, the rare association of AS (A), mental retardation (M), midface hypoplasia (M), and erythrocyte elliptocytosis (E) has been recognized as another X-linked contiguous gene deletion syndrome (AMME) (65).
Immunohistologic Study of Renal, Epidermal, and Other Extrarenal Basement Membranes
As early as 1972, Spear and Slusser (10) speculated that the primary defect in AS could involve basement membrane collagen. This hypothesis was subsequently confirmed by a series of immunohistochemical, biochemical, and genetic studies. McCoy et al. and Olson et al. observed that glomeruli from patients with AS failed to bind human anti-GBM antibodies from patients with Goodpasture (GP) syndrome, a finding suggesting that an antigen normally present in the GBM was absent in these patients (66,67). This observation was confirmed by different studies using the same type of sera or monoclonal or polyclonal antibodies directed against the GP antigen later identified as the NC1 domain of α3(IV) (31). Overall, most male patients showed absent or reduced binding of the antibodies to the GBM, but this phenomenon was not universal, a finding confirming the heterogeneity of AS. Intrafamilial concordance of immunolabeling in male patients was consistently observed (66), and lack of GBM labeling was correlated with the severity of renal disease and GBM splitting (68). Variable results have been found in female patients, with absent, reduced, or even normal immunolabeling in female patients whose affected male relatives lacked the GP antigen (68).
Parallel to the studies on the GP antigen, the Minneapolis group showed that the serum (FNS1) of an AS patient who had developed anti-GBM antibodies after renal transplantation recognized an antigen more widely distributed than the GP antigen and notably present in the epithelial basement membrane (EBM) (28). This antigen was shown to be absent in the GBM and EBM of most AS patients and to have a mosaic distribution in related heterozygous female patients, suggesting that it belongs to a new type IV collagen chain, the possible substrate of AS. This hypothesis was confirmed by the demonstration that FNS1 recognizes the NC1 domain of the α5 chain of type IV collagen.
With the modern availability of specific monoclonal or polyclonal antibodies, immunohistologic analyses of the basement membrane distribution of the different chains of type IV collagen have been performed in numerous AS patients (7,12,29,40,42,48,51). Within the kidney, abnormal distribution of the α5(IV) chain, the product of the mutated COL4A5 gene, is observed in approximately 75% of the patients: the antigen is absent in the GBM and the distal TBM in male patients and has a reduced or discontinuous distribution in related female patients (Fig. 13.11). This mosaic pattern of GBM distribution of the α5(IV) chain in female patients is consistent with the X-linked transmission of the disease. Random inactivation of one of the two X chromosomes in heterozygotes may explain different degrees of severity of the disease, in correlation with normal, absent, or discontinuous expression of the antigen along the GBM. Interestingly, the α5(IV) defect is associated with the co-absence of the α3 and α4(IV) chains that normally participate in the formation of the α3.α4.α5(IV)-α3.α4.α5(IV) network. Similarly, the α6(IV) chain, normally part of the α5.α5.α6(IV) network, is not detected in the Bowman capsule and collecting duct basement membranes. Exceptions to this typical pattern have been reported in a few patients, characterized by the absence of detectable α5(IV) contrasting with the persistence of discrete α3(IV) and α4(IV) GBM expression (12,29). In about one third of X-linked AS patients, no significant changes in the renal distribution of the α(IV) chains are detected by conventional immunohistologic methods. Results of immunolabeling are concordant within families, and correlation of abnormal labeling with the severity of the clinical and pathologic phenotype is usually observed.
In contrast to the glomerular loss of the α3.α4.α5(IV) network, the α1(IV) and α2(IV) chains, normally confined to the mesangium and the subendothelial aspect of the GBM, are present throughout the entire width of the GBM where they are codistributed with collagen types V and VI and laminin α2 (see Fig. 13.11A) (29,48,69).
In most X-linked AS, the EBM distribution of α5(IV) (and α6[IV] when performed) is abnormal, being absent in male and segmental (or discontinuous) in female patients (see Fig. 13.11H and I) (7,29,42,70,71). GBM and EBM changes are always concordant in male patients, a finding confirming the high diagnostic value of immunohistologic examination of the skin, a technique that had become the initial diagnostic approach in several laboratories. However, approximately 25% of male patients have normal α5(IV) staining of the skin (and the kidney) when examined by standard fluorescence microscopy, and normal results in female patients do not allow any definitive diagnostic conclusion because of the segmental distribution of the antigen.
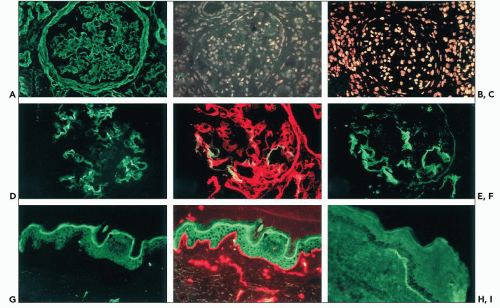 FIGURE 13.11 X-linked Alport syndrome. Immunofluorescence distribution of type IV collagen chains in the renal (A-F) and epidermal (G-I) basement membranes of patients affected with X-linked Alport syndrome. In a male patient (A-C), no α3 or α5(IV) labeling (B, C), contrasting with the strong linear α1(IV) labeling of the GBM (A). In a female patient (D-F), discontinuous α5(IV) GBM labeling (D, F), with strong α2(IV) labeling of α5(IV)-negative GBM segments (E). No EBM α5(IV) staining in a male patient (G), with preservation of α2(IV) labeling (H). Segmental α5(IV) labeling in a female patient (I). (α1, α3, α5: FITC; α2: Texas red.) |
The EBM expression of α5(IV), used as a marker of X inactivation, has been found to be correlated with the severity of the renal disease in female patients and proposed as a marker for predicting patient outcome (72). However, this finding was not confirmed by the study of Massella et al. (73), in which no correlation was observed between the severity of the glomerular involvement in female patients and the extent of EBM α5(IV) staining. Accordingly, X inactivation can vary widely between different tissues from the same individual (74).
Few studies have focused on the extrarenal basement membrane other than the EBM, because the involved tissues in the eye, ear, and esophagus are not readily accessible. Lens capsules from three patients with AS with anterior lenticonus were examined. The distribution of the α3(IV) to α5(IV) chains was normal in one patient, whereas all three chains (as well as α6[IV] in one case) were absent in the others (26,44). The α3(IV) and α5(IV) chains were also found to be absent in the basement membrane overlying the basilar membrane in two X-linked AS patients (24). The α5(IV) and α6(IV) chains, which are normally expressed in the esophageal smooth muscle cell basement membrane, are absent in esophageal tumors from patients with AS and diffuse esophageal leiomyomatosis (75). Curiously enough, a COL4A6 transcript was strongly expressed by the tumor cells. An additional striking feature was the absence of the β1 chain of laminin in tumor BM and the lack or uneven expression of the α5 integrin subunit by tumor cells (75).
Genetics
After decades of discussion, reexamination of large pedigrees led to the conclusion that AS was genetically heterogeneous, with high frequency of X-linked dominant transmission (50). Linkage studies using polymorphic markers located on chromosome X allowed the localization of the gene at Xq22 (78). As soon as it was identified by two groups in Finland (36,37), the gene COL4A5 became the candidate gene for X-linked AS and the first COL4A5 mutations were identified in three Alport kindreds (79). Today more than 500 different mutations have been reported (31,51,80,81) and collected in a database (82). They range from small or large rearrangements or deletions to small point mutations, dispersed all over the gene. Most of these rearrangements are unique. According to different groups, de novo mutations occurred in 11% to
15% of patients. No mutations have been detected in only the COL4A6 gene (i.e., sparing the COL4A5 gene) located in tandem on the same chromosome, which is not surprising since the chain α6(IV) is not expressed in the GBM.
15% of patients. No mutations have been detected in only the COL4A6 gene (i.e., sparing the COL4A5 gene) located in tandem on the same chromosome, which is not surprising since the chain α6(IV) is not expressed in the GBM.
Gross COL4A5 rearrangements (mainly deletions) have been identified in 5% to 15% of the families. The size and location of the deletions vary from case to case, involving from one exon to the entire length of the gene. They are intragenic or extend to nearby regions of the chromosome, possibly involving the adjacent COL4A6 gene. In some intragenic deletions, an open-reading frame is conserved, allowing the synthesis of a shortened COL4A5 protein, whereas frameshift deletions lead to more severe truncation of the protein. Insertions, duplications, or complex rearrangements of the gene have also been described.
Many small mutations in the COL4A5 gene have been reported. They are distributed throughout the gene. Single base mutations leading to amino acid substitution represent about 40% of the small mutations. Most of them are missense mutations in glycine codons in the collagenous domain of COL4A5. They are considered pathogenic because they disrupt the Gly-X-Y repeats, altering the folding of the collagen triple helix and leading to posttranscriptional modifications of the chains. Other missense mutations affect conserved amino acid residues, especially cysteine, in the NC domain of the α5 chain. Nonsense mutations, small deletions or insertions, and splice-site mutations leading to frameshift and premature stop codons result in truncated proteins that lack the carboxy terminal NC domain of the chain.
No “hot spot” of mutation has been found. However, a few mutations have been detected in more than one family. For example, one point mutation has been identified in nine independently ascertained AS kindreds in the United States. However, genealogic studies and haplotype analysis demonstrated that affected patients are descendants of a common Mormon ancestor (83). Similarly, the high frequency of AS in French Polynesia is caused by a founder mutation, a tandem duplication of 35 COLA5 exons (17).
The single-strand conformation polymorphism (SSCP) method used for screening the 51 exons of the COL4A5 gene allowed the identification of mutations in 45% to 65% of families (17,80). The mutation detection rate reaches 80% to 90% when more sensitive methods are used: direct DNA sequencing or reverse transcriptase-polymerase chain reaction (RT-PCR) and direct sequencing using mRNA from leukocytes, hair roots, or cultured skin fibroblasts (84,85,86,87). New-generation sequencing protocols will allow rapid and less expensive identification of mutations (88).
Phenotype-Genotype Correlations
Two large European and the U.S. cohorts assembling 195 and 175 families, respectively, have been studied to establish phenotype-genotype correlations in male patients (401 and 681 in respective cohorts) affected with X-linked AS (51,81). Both studies demonstrated the uniform severity of large rearrangements of the COL4A5 gene, nonsense mutations, and deletion or insertions that change the reading frame of the gene, resulting in the synthesis of a truncated α5(IV) chain without the NC1 domain. These mutations confer on affected male patients a probability of developing ESRD before the age of 30 years of 90%, with a 50% renal survival rate at 20 years (51), and an average age at onset of ESRD of 25 years (81). In contrast, phenotypic heterogeneity is seen in the group of patients with missense mutations. In them, the probability of developing ESRD before the age of 30 is 50%, with a 50% survival rate at 32 years, and a mean age at ESRD of 37 years. In other words, both juvenile-type and adult-type AS may be observed in patients with missense mutations, and intrafamilial variability in the rate of progression of the disease is possible. In patients with splice-site mutation, the severity of the renal disease is intermediate with a risk of developing ESRD before the age of 30 of 70% and an average age at ESRD of 28 years. A meta-analysis of phenotype-genotype correlations based on 44 publications suggested a more severe renal phenotype in patients with 3’compared with 5’glycine missense mutations and in patients with splice donor compared with splice acceptor mutations (80). This correlation was not observed in the US cohort, in which mutations positioned at the 5′ end of the gene were found to be associated with earlier age at ESRD onset (81).
Similarly, missense mutations are associated with a lower and later incidence of hearing impairment (51,81). They confer a 50% risk of hearing defect at age 20, whereas all other types of mutations lead to the same risk at age 10. Absence of deafness is observed in some kindreds with COL4A5 missense mutations; this finding shows that “hereditary nephritis without deafness” is a variant of AS. Interestingly, Samoyed dogs with a nonsense mutation in COL4A5 leading to the absence of the α5(IV) chain have no detectable hearing defect. In these studies, no significant correlation was observed between the finding of maculopathy and the type of mutation. Conversely, the frequency of lenticonus is significantly higher in patients with large deletions or small mutations, resulting in premature stop codons (51).
In girls and women, no significant correlation was observed between genotype and phenotype, and no correlation was established between the phenotype in girls and women and the severity of the renal disease in male relatives or offspring (52). Early prognosis of the disease in AS carriers is not possible.
An interesting observation has been made in patients suffering from AS associated with diffuse esophageal leiomyomatosis. They all have large deletions encompassing the 5′ end of both COL4A5 and COL4A6 genes and the regulatory intergenic region; the deletions, in COL4A6, are limited to exons 1, 1′, and 2 (63). Interestingly, AS patients with larger deletions in COL4A6 do not develop tumors (63).
Autosomal Recessive Alport Syndrome
In 1985, the genealogic analysis of 41 kindreds by Feingold et al. (50) suggested that AS was transmitted as an autosomal recessive trait in about 10% of cases, a frequency confirmed by further studies. Homozygous or compound heterozygous mutations in COL4A3 or COL4A4 have now been reported in several kindreds (34,35,89,90,91).
Clinical features are identical to those observed in X-linked AS. Autosomal recessive inheritance is suggested by the severity of the disease in young female patients, consanguinity in the family, or the presence of microscopic hematuria in the father of a male patient. Progression to ESRD is constant in both sexes and usually occurs before the age of 30. A few patients reach ESRD around 10 years of age or as late as 44 years. Hearing impairment is nearly constant, and ocular lesions may or may not be associated. Ultrastructural
changes of the GBM identical to those in X-linked AS are present. Immunohistochemical distribution of type IV collagen chains may be normal, but a peculiar pattern of renal and skin distribution of the α3(IV) to α6(IV) chains is observed in most patients (92). It is characterized by the co-absence of the α3, α4, and α5 chains in the GBM, replaced by the α1.α1. α2(IV) network, whereas the α5(IV) and the α6(IV) chains are present in the Bowman capsule and collecting duct basement membranes, and in EBM, as normally seen (Fig. 13.12). Taking into account the normal distribution of the type IV collagen networks, these findings indicate that defects in COL4A3 or COL4A4 result in the defective expression of the α5(IV) chain only in those basement membranes in which the three chains are associated within the α3.α4.α5(IV)-α3.α4.α5(IV) network.
changes of the GBM identical to those in X-linked AS are present. Immunohistochemical distribution of type IV collagen chains may be normal, but a peculiar pattern of renal and skin distribution of the α3(IV) to α6(IV) chains is observed in most patients (92). It is characterized by the co-absence of the α3, α4, and α5 chains in the GBM, replaced by the α1.α1. α2(IV) network, whereas the α5(IV) and the α6(IV) chains are present in the Bowman capsule and collecting duct basement membranes, and in EBM, as normally seen (Fig. 13.12). Taking into account the normal distribution of the type IV collagen networks, these findings indicate that defects in COL4A3 or COL4A4 result in the defective expression of the α5(IV) chain only in those basement membranes in which the three chains are associated within the α3.α4.α5(IV)-α3.α4.α5(IV) network.
There is a broad spectrum of phenotypes in heterozygous carriers. Some are completely asymptomatic, whereas others present with persistent or intermittent microscopic hematuria associated with the thin GBM in the few cases examined (34,35), that is, with the typical clinicopathologic features of “familial benign hematuria” (93). Conversely, progression to renal failure has been observed in a few heterozygous carriers (35). This raises the question of the advisability of using relatives as living donors for kidney transplantation in recessive AS.
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
Pauci-Immune and Antineutrophil Cytoplasmic Autoantibody-Mediated Crescentic Glomerulonephritis and Vasculitis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


