TABLE 25.1 Main etiologic and pathogenetic factors responsible for TIN | ||||
|---|---|---|---|---|
| ||||
value of only 38%. The same authors also examined consecutive patients with WBC in the urine who did not have interstitial nephritis. Four of these patients had urinary eosinophils greater than 1%. Eosinophiluria is not uncommon in secondary forms of interstitial nephritis, particularly in those that are associated with crescentic glomerulonephritis (vasculitis).
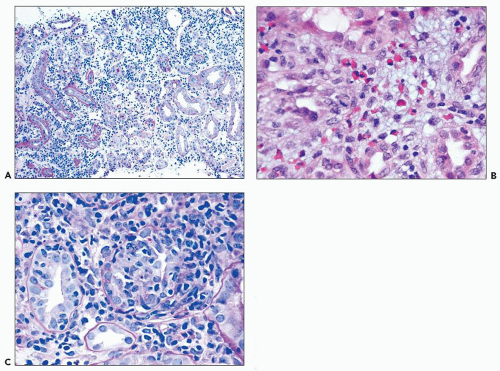 FIGURE 25.1 Interstitial nephritis in a 66-year-old patient who did not have any identifiable underlying etiology but had peripheral eosinophilia. A: Interstitial mononuclear cell infiltrate with edema. (PAS, ×100.) B: Focally large numbers of eosinophils were present in the interstitium. (H&E, ×400.) C: In several foci, the inflammatory cells infiltrated the tubular epithelium (tubulitis). (PAS, ×600.) |
common in drug-induced cases, but their absence does not exclude a drug-induced form of interstitial nephritis (21). After a few days or weeks have elapsed, a variable accumulation of plasma cells and histiocytes may be present (Fig. 25.3). Although not a common component of acute TIN, granuloma formation may occur in drug reactions, sarcoidosis, and idiopathic forms (Fig. 25.4) (3). If many plasma cells are seen, the diagnosis of IgG4-related interstitial nephritis should be considered, and an immunostain for IgG4 should be performed (13) (Fig. 25.5).
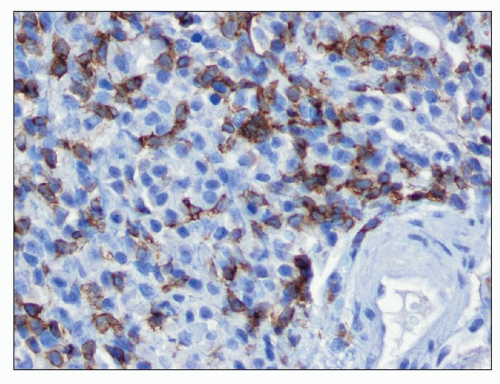 FIGURE 25.2 Immunohistochemistry reveals many T cells in the interstitial inflammatory cell infiltrate in this biopsy from a patient with Sjögren syndrome. (Immunoperoxidase with an anti-CD3 antibody, ×400.) |
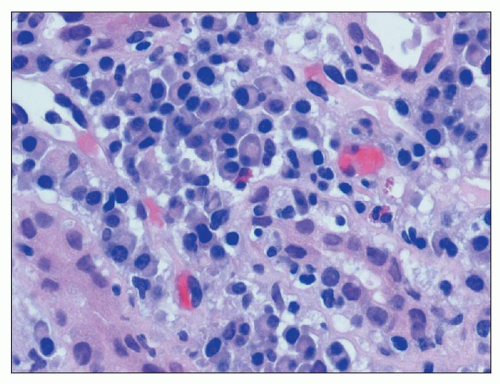 FIGURE 25.3 Many plasma cells in an acute and chronic interstitial nephritis, in a patient with Sjögren syndrome. (H&E, ×600.) |
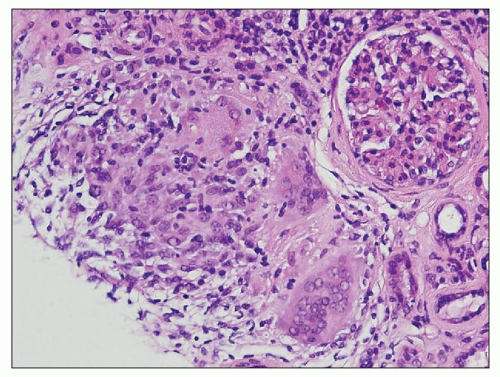 FIGURE 25.4 Granulomatous interstitial nephritis. Well-defined epithelioid granuloma with giant cells in the renal biopsy of a patient with sarcoidosis. (H&E, ×100.) |
older persons, they are unrelated to the primary tubulointerstitial process and reflect aging, associated hypertension, or both.
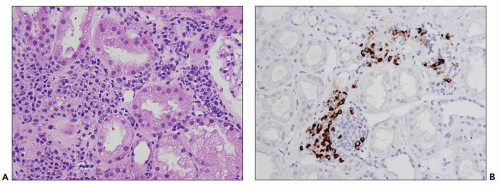 FIGURE 25.5 IgG4-related interstitial nephritis. A: Numerous plasma cells are seen in interstitial inflammatory cell infiltrates. (H&E, ×200.) B: Immunohistochemistry shows multiple IgG4-positive plasma cells. (Immunoperoxidase, ×200.) |
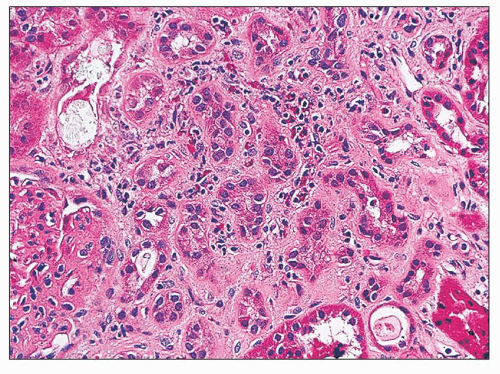 FIGURE 25.6 Active appearing interstitial inflammatory cell infiltrate with eosinophils in the background of interstitial fibrosis and tubular atrophy in a patient with a long history of gout and multiple medication use. (H&E, ×200.) |
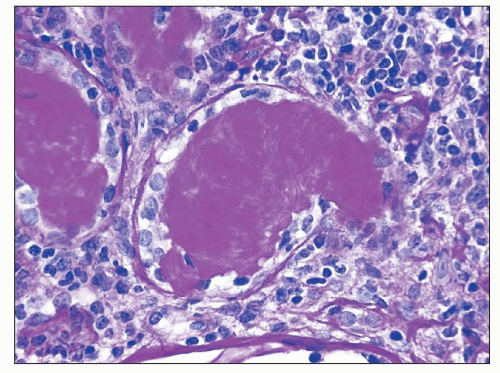 FIGURE 25.7 Tubular rupture with expulsion of Tamm-Horsfall protein from the tubule into the interstitium. Note the interstitial inflammatory cell infiltrate around the Tamm-Horsfall protein. This is a nonspecific finding that can occur in any renal injury with tubular disruption and secondary interstitial Tamm-Horsfall protein deposits. (PAS, ×400.) |
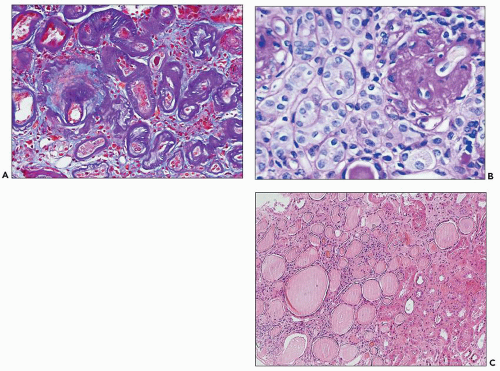 FIGURE 25.8 Different histologic appearances of atrophic tubules. A: Prominent thickening of the TBM in “classic” atrophic tubules highlighted by double PAS-trichrome stain (28). Note violet appearance of the TBM and blue interstitial collagen between the atrophic tubules. (PAS-trichrome double stain, ×200.) B: Endocrine-type atrophic tubules surrounding a sclerotic glomerulus. In this type of atrophic tubule, the basement membrane is thin and the epithelium is simplified with no or only very narrow lumen. These tubules resemble endocrine glands. (PAS, ×400.) C: Thyroidization of tubules in a scarred area of the renal cortex. Thyroid-type atrophic tubules have flattened epithelium and PAS-positive proteinaceous filling the lumen, resembling thyroid follicles. (H&E, ×100.) |
including tubular epithelial cells and endothelial cells, also contribute to the extracellular matrix deposition by producing fibronectin, type IV collagen, and a variety of other matrix proteins (31). Interstitial fibrosis and tubular atrophy are cardinal features for the diagnosis of chronic TIN because inflammatory cells may be scarce or absent.
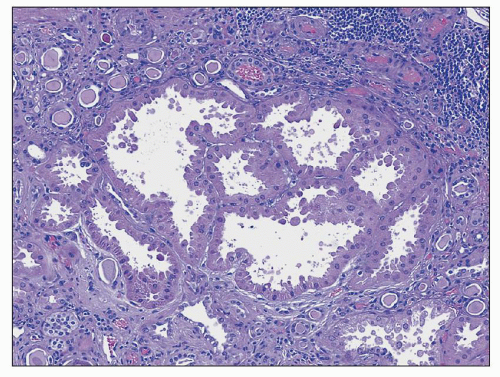 FIGURE 25.9 It is common to see large hypertrophic tubules with thick, hypertrophic epithelial lining in any type of advanced chronic renal injury. (H&E, ×100.) |
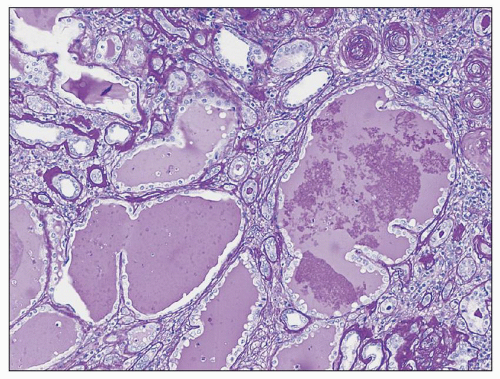 FIGURE 25.10 Microcystic dilation of tubules with scalloped outline. Such tubules can be seen in any kind of chronic tubulointerstitial injury. (PAS, ×100.) |
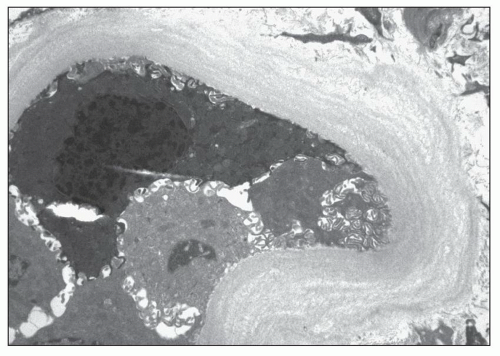 FIGURE 25.11 Thickened lamellated basement membrane of an atrophic tubule. (Uranyl acetate and lead citrate, ×3000.) |
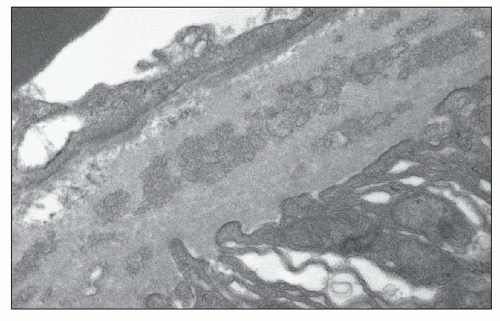 FIGURE 25.12 Deposits of granular to microspherical material in the TBM is a common finding in atrophic tubules. Under low magnification, these structures may be misinterpreted as electron-dense immune-type deposits in the TBM. (Uranyl acetate and lead citrate, ×20,000.) |
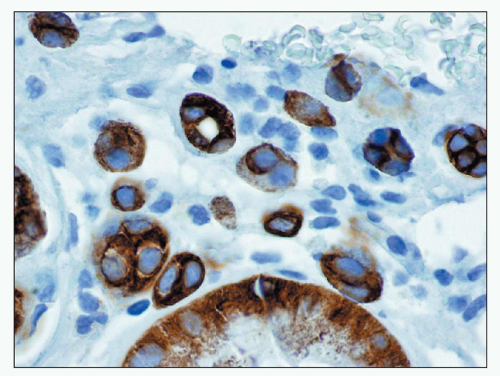 FIGURE 25.13 Scattered cytokeratin-positive cells are commonly found in the fibrotic renal interstitium. It is theoretically possible that these cytokeratin-positive cells represent cells undergoing epithelial-tomesenchymal transformation. (Immunoperoxidase, ×600.) |
TABLE 25.2 Causes of granulomatous interstitial nephritis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
and clinical findings. It is possible that several mechanisms of action are at work in an individual patient.
have shown a picture of interstitial edema with variable numbers of mononuclear cells, accompanied by variable degree of acute tubular injury (76,82,83). Granulomas may be seen in some cases (84). No immunoglobulins or complement have been seen with immunofluorescence techniques.
reported (113). A few investigators described fibrillar deposits along distal convoluted tubules and in glomerular epithelial cells (102,105), but the relevance of these fibrils is unclear, and they may merely represent procollagen.
vancomycin combination therapy (142). Some patients have an associated rash and eosinophilia, suggesting a hypersensitivity reaction. In addition to these adverse renal effects, in some cases, patients have an anaphylactoid reaction to the drug, with generalized flushing—the so-called red man syndrome.
following 4.2 ± 0.7 days’ mean duration of treatment, and the Scr returned to normal following withdrawal of NSAIDs. They estimated the incidence of AKI, defined as the number of recognized cases per inpatient days of therapy, and found it to be 0.001, 0.0003, 0.0001, and 0.0001 for indomethacin, ibuprofen, zomepirac, and sulindac, respectively (166). The incidence may be overestimated in some of these studies because hospitalized patients already represent a selected population and may have risk factors for NSAID toxicity (see later).
TABLE 25.3 Nonsteroidal anti-inflammatory drugsa | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
impairment associated with chronic hepatic failure (168). In addition, cirrhotic patients have enhanced renal PG-E production, which is vasodilatory. Administration of NSAID to these patients may profoundly reduce renal blood flow and GFR (182).
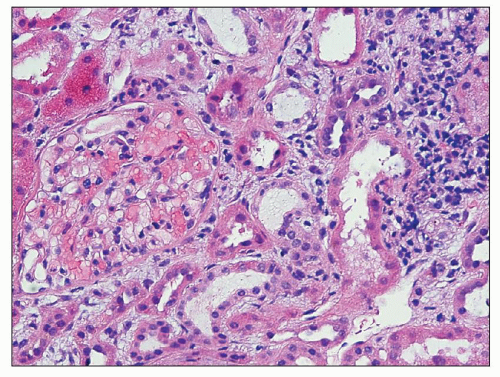 FIGURE 25.14 Mild interstitial edema and inflammation, associated with acute tubular injury, in a patient following NSAID administration. The glomerulus is unremarkable. This patient had nephrotic syndrome and acute renal failure. The renal failure and proteinuria reversed after discontinuation of NSAIDs. (H&E, ×200.) |
water and salt reabsorption (168). As mentioned earlier, NSAIDs do not alter the GFR in healthy, euvolemic individuals (179). In contrast, in conditions where the systemic hemodynamic conditions are compromised, NSAIDs may have a deleterious effect on the renal circulation. Owing to the inhibition of COX, the synthesis of vasodilatory PGI2 and PGE2 is diminished, and severe, unbalanced renal vasoconstriction may develop, resulting in AKI (168,195).
nonphenacetin combined analgesics are truly associated with nephropathy (213).
abuse-associated analgesic nephropathy, and it is not entirely clear whether non-phenacetin-related cases have the same capillary calcification. This capillary sclerosis increases in intensity toward the pelvic-ureteric junction, is most prominent in the proximal ureter, and then gradually decreases (224). At a more advanced stage (in early stages of papillary necrosis), the capillary sclerosis involves the peritubular capillaries in the papilla and inner medulla. The ascending loop of Henle also exhibits a substantially thickened basement membrane, but the basement membranes of the collecting ducts, descending loop of Henle, and vasa recta are not affected or are only mildly affected. The thickened basement membranes are PAS positive and contain lipid as well as calcium deposits (Fig. 25.16). Ultrastructurally, this basement membrane thickening consists of numerous thin layers of basement membrane material (Fig. 25.17), which probably forms as the result of repeated injury of the capillary endothelium and the epithelium of the thin limb of Henle (224,228). Early on, these changes are confined to the central part of the inner medulla, but as the disease progresses, the affected small foci become confluent and may involve the entire inner medulla.
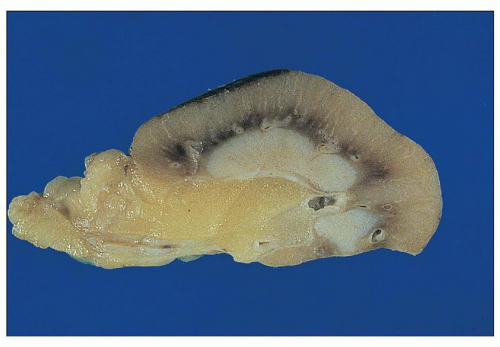 FIGURE 25.15 Portion of a kidney with advanced analgesic nephropathy. Note the pale gray-white papilla, representing papillary sclerosis/necrosis. |
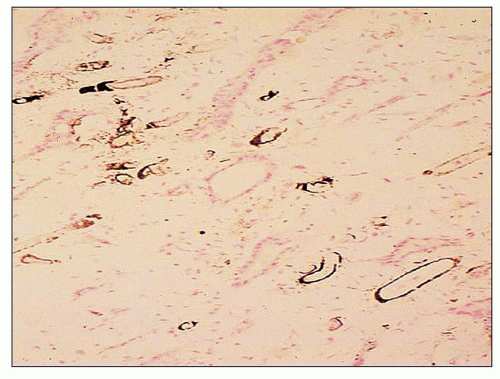 FIGURE 25.16 Calcium deposits in the basement membranes of the vasa recta in the renal papilla in analgesic nephropathy. (Von Kossa, ×200.) |
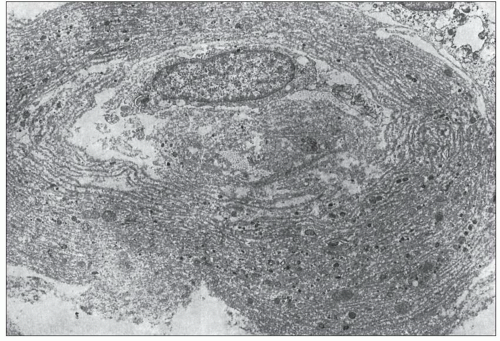 FIGURE 25.17 Electron micrograph of a capillary obtained by biopsy of the renal pelvis of a patient who had abused analgesics. Numerous new basement membrane lamellae have been formed. (×7050.) (From Mihatsch MJ, et al. The morphologic diagnosis of analgesic (phenacetin) abuse. Pathol Res Pract 1979;164:68.) |
early and intermediate forms. The cortical changes consist of tubular loss and tubular atrophy with interstitial fibrosis and a varying degree of interstitial infiltration of chronic inflammatory cells (Fig. 25.20). Lipofuscin accumulation is frequently noted in the epithelium of atrophic tubules. These are nonspecific changes and cannot be reliably differentiated from other forms of chronic tubulointerstitial injury. It appears that the necrotic papilla, in some ways analogous to obstructive nephropathy, is responsible for the cortical changes. This is also supported by the fact that the columns of Bertin are often spared.
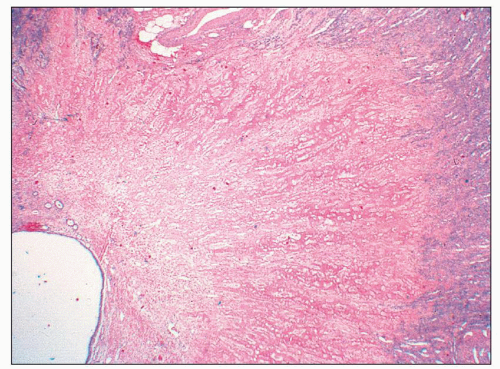 FIGURE 25.18 Low-magnification picture taken from the specimen shown in Figure 25.15. In analgesic nephropathy, typically there is no inflammatory reaction around a necrotic/sclerotic papilla. The ghost structure of the renal papilla is still recognizable. (H&E, ×10.) |
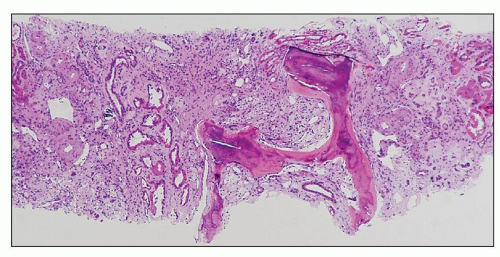 FIGURE 25.19 Bone formation in a necrotic papilla from a patient who had used analgesics for many years. (H&E, ×140.) |
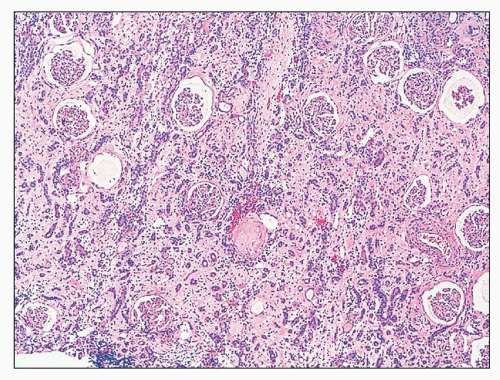 FIGURE 25.20 Interstitial fibrosis and tubular atrophy in the cortex of a kidney with analgesic nephropathy. Note that most glomeruli in this section are preserved; only scattered sclerotic glomeruli are seen. (H&E, ×40.) |
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Glomerular Diseases With Organized Deposits
Glomerular Diseases With Organized Deposits
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


