prognosis and therapy. Current clinical classification is based primarily on the patient’s age, clinical presentation, and anticipated treatment rather than etiology. This is because there is an urgency to initiate treatment usually well before etiologic diagnosis is typically available. Once etiology is determined, treatment can be adjusted, if needed. However, the lack of etiologic information at the time of presentation is not the only barrier for the implementation of a purely etiologic classification. Etiologic diagnoses based on some of the key laboratory parameters, such as ADAMTS13 activity, may not be appropriate for the critical initial decision needed for treatment. For example, abnormally low ADAMTS13 activity does not identify all patients who will respond to plasma therapy (9). There is no uniformly accepted classification of TMAs in use. The clinical classification discussed below has four major diagnostic categories (Table 18.1), that is, classic HUS, atypical HUS (aHUS), TTP, and “other” TMAs. Because of historical as well as practical reasons, this classification also includes the secondary forms of aHUS (i.e., those other than familial and idiopathic forms) and the secondary forms of TTP (i.e., those other than congenital and idiopathic forms), under the aHUS and TTP categories, respectively. It should be emphasized that clinical recognition of various subsets, including the secondary forms, within the major categories at the onset of the disease is often difficult or impossible. Therefore, the rule of thumb is that at the time of presentation, patients are presumptively assigned into one of the four major diagnostic categories to guide initial treatment. MAHA, thrombocytopenia, and renal impairment with a diarrhea prodrome caused by Shiga toxin (Stx)-producing Escherichia coli (E. coli) are the diagnostic hallmarks of classic HUS. Most of these patients are children under the age of 5. However, adults with a history suggesting Stx-producing E. coli (STEC) infection are also treated with plasma exchange because the etiology cannot be certain when decision about plasma exchange needs to be made (12). Diagnosis of aHUS relies on the presence of MAHA, thrombocytopenia, and renal impairment, and exclusion of associated disease(s), Stx-associated HUS, and TTP (11). In most instances, however, aHUS cannot be distinguished from TTP at the time of presentation. Until recently, this had only limited therapeutic ramifications since the first line of treatment for aHUS has been plasmapheresis, similar to that of TTP. If additional clinical and laboratory data do not confirm the presumptive clinical diagnosis, the diagnosis is revised and treatment is readjusted, as needed. Those patients who fulfill the diagnostic criteria of TTP (MAHA, thrombocytopenia, with or without renal or neurologic abnormalities, and exclusion of systemic infections and other causes of TMA) are diagnosed as such and treated accordingly with plasmapheresis (9). The majority of patients diagnosed with TTP are adults; however, children without renal impairment can also be diagnosed with TTP. Those cases that do not fit into any of the aforementioned categories will be classified as “other TMAs” with a designation of associated disease(s) or condition(s). The current definition of aHUS limits the subgroups within the aHUS category to those familial and idiopathic forms (13), and all secondary forms are delegated to the “other TMA” category and designated as such (Tables 18.2 and 18.3). Some investigators further narrow the idiopathic group of aHUS to only those cases with complement abnormalities (11) and refer to them, along with the familial forms with complement abnormalities, as “primary” HUS or “complement”-HUS. HUS caused by Streptococcus pneumoniae is classified as a form of aHUS by some and, as a separate category, often designated as S. pneumoniae-associated HUS by others (14,15,16). The term TTP is applied to only those cases in the congenital and idiopathic groups, while all other cases formerly considered as various subgroups of TTP (“secondary” TTPs) are now designated as “TMAs” or “other TMAs.” In this modified classification, the clinical diagnoses are better aligned with etiology in the HUS and TTP groups; however, the “other TMA” group is still quite heterogeneous. To avoid potential confusion due to terminology, some experts prefer the all-inclusive morphologic term TMA instead of HUS and TTP, while others use the hybrid term HUS/TTP.
TABLE 18.1 Clinical classification of TMA | |
|---|---|
|
America and Europe, and develops in isolated cases or as outbreaks occurring mostly in the summer (17). Although the designation “epidemic” HUS is also used for the classic form of HUS, the majority of cases are indeed sporadic. Furthermore, gastroenteritis is a common trigger for the atypical form of the disease, and therefore the term D+ HUS can be misleading. In North America and Western Europe, most of the classic forms are associated with O157:H7 serotype of Shiga toxin-producing E. coli (STEC) infection (18,19,20,21). However, many other serotypes of STEC have also been linked to classic HUS (20,22,23,24,25,26). Infection with Shiga toxin-producing Shigella dysenteriae serotype 1 has been a common cause of classic HUS in developing countries in Asia (27) and Africa (11,28,29,30), but not in industrialized countries (31). The annual incidence of classic HUS is estimated to be 21 per 1 million with a peak incidence in children under the age of 5 years (61 per 1 million) and the lowest rate in adults in the age group of 50 to 59 years (5 per 1 million) (32). TMA in the classic form due to STEC infection is most often confined to the glomeruli, with a consequently good prognosis.
TABLE 18.2 Classification of HUS and TTPa | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
TABLE 18.3 Thrombotic microangiopathies other than classic and aHUS and TTPa | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
implementation of MAHA and thrombocytopenia as the new diagnostic criteria for TTP in a proper clinical setting (86). In a recent publication relevant to the current clinical practice, the diagnostic pentad of TTP was only present in 3 out of 69 patients (5%) with severe ADAMTS13 deficiency at the time of presentation (9). This change in the diagnostic criteria of TTP over time has further narrowed the differences between HUS and TTP. However, distinction between TTP and HUS has gained ground with the recognition that a significant proportion of patients with TTP have very low ADAMTS13 activity, while many of the patients with the atypical form of HUS have complement regulatory factor abnormalities. Although these findings proved to be very helpful to our understanding of the pathogenesis, abnormal ADAMTS13 activity is not diagnostic of TTP since patients with other conditions, such as systemic infections, might also have significantly diminished ADAMTS13 activity. Furthermore, the activity is normal in approximately 50% of cases with idiopathic TTP, and abnormal ADAMTS13 activity does not identify all patients who will benefit from plasmapheresis (9,72). In regard to complement regulatory factor abnormalities, since penetrance is only 50% in those family members who carry the mutations, the mutations seem to be risk factors rather than unique causes of the disease. In addition, mutations are only identified in approximately 50% of patients with idiopathic aHUS, further limiting the diagnostic and prognostic utility of the test. Finally, since the prevalence of the complement regulatory factor mutations in the general population is unknown, the finding of a mutation in an asymptomatic patient is difficult to interpret.
of all patients with HUS during this outbreak was 44 years, and among those 101 pediatric patients (i.e., 17 years of age or younger), the median age was 11 years (75). It remains unclear whether the atypical age distribution in this most recent outbreak reflects the pattern of sprout consumption (the most likely vehicle for the infection) or is attributable to the specific properties of the serotype (O104:H4) of the causative E. coli strain (75).
pathogenic hybrid organism possessing features common to both enteroaggregative E. coli (EAEC) and STEC (121,122,123). EAHEC strains have evolved from EAEC that cause watery diarrhea in children and travelers’ diarrhea, by acquiring genes for Stx2a and antibiotic resistance (121,122). Except for Stx2a, no other EHEC-specific virulence markers including the locus of enterocyte effacement (LEE) are present in EAHEC strains. EAHEC O104:H4 colonizes the bowel through aggregative adherence fimbrial pili encoded by the EAEC plasmid. The aggregative adherence fimbrial colonization mechanism substitutes for the LEE functions for bacterial adherence and delivery of Stx2a into the colonic mucosa, ultimately resulting in HUS (25,121,122).
disorders, and pregnancy (185). In some, and perhaps a significant proportion of patients, TTP can be mimicked by AIDS-related conditions, comorbidities, and multiple drugs in use. Also, the possibility of a specific HIV-associated form of TMA (other than HUS or TTP) has been hypothesized (190). Findings that may support the existence of such form of TMA include the presence of endothelial dysfunction in association with HIV infection (191), and also human herpesvirus 8 infection involving endothelial cells, common in HIV-infected patients (192). A number of other factors have also been implicated as possible triggers or predisposing factors for TMA in HIV-infected patients. These include CMV infection, cryptosporidiosis, and AIDS-related malignancies (180,181,193).
in patients treated with anti-VEGF agents (54). This is also supported by experimental evidence showing that local genetic ablation of VEGF from podocytes in the mouse causes TMA similar to that seen in humans (54,219). TMA has also been described in association with various immunotoxins, such as Combotox (225), antibodies including Apolizumab (Hu1D10) (226), as well as the tyrosine kinase inhibitor Imatinib (Gleevec) (227).
syndrome during pregnancy was associated with a significant fetal loss (50%), an incidence substantially higher than that seen in the noncongenital forms of the disease (276,279). The presence of complement abnormalities during pregnancy is also a risk factor for fetal loss and preeclampsia; however, the incidence of such complications appears to be relatively low (4.8% and 7.7%, respectively) (61,280). There is a significant overlap between the clinical and laboratory manifestations of eclampsia-preeclampsia, pregnancy-associated TTP, and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets) (279). Since these conditions require different therapies, difficulty in differentiating these conditions from each other remains a significant clinical problem. Although abnormal ADAMTS13 activity is not specific for TTP, severe deficiency or absence of ADAMTS13 activity favors TTP over HELLP syndrome and preeclampsia-eclampsia (281).
TABLE 18.4 Renal morphologic features of TMA | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
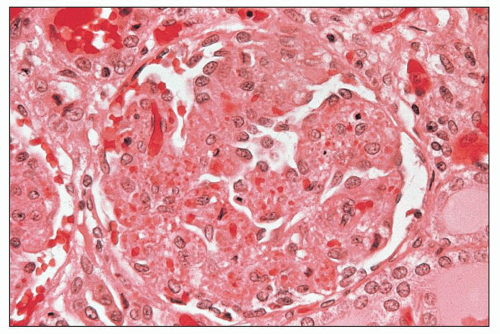 FIGURE 18.1 Hemolytic-uremic syndrome, classic form. The glomerulus is slightly hypocellular, and most of the glomerular capillary lumina are closed due to thickening of the capillary walls. Red blood cells and fragmented red blood cells are seen in the mesangial areas. The specimen is from a child with STEC infection. This type of glomerular change is typical during the acute stage of the disease. (H&E) (Courtesy of Dr. Vivette D’Agati.) |
of polymorphonuclear leukocytes in the glomerular capillaries can be seen in some cases; this feature may be prominent in patients with classic HUS (311). Capillary thrombi, endothelial swelling, and congestion were identified as the typical glomerular findings in patients with severe classic HUS (311). The glomerular capillary thrombi in TTP are usually not very extensive (Fig. 18.8). On occasion, the majority of the glomeruli may be affected, but in general, the glomerular changes in TTP are usually not as dramatic as in HUS.
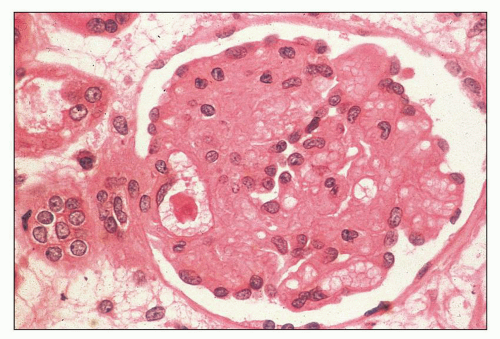 FIGURE 18.2 Hemolytic-uremic syndrome, atypical form. “Bloodless” glomerulus. The glomerular capillary walls are thickened, and the mesangial areas blend with the capillaries. (H&E) (From Kern WF, et al. Atlas of Renal Pathology. W.B. Saunders Company, Philadelphia, 1999, with permission.) |
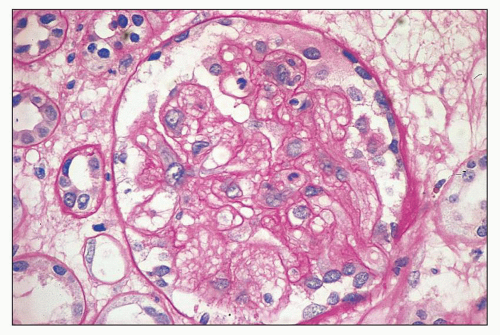 FIGURE 18.3 Hemolytic-uremic syndrome, atypical form. The mesangial areas of the glomerulus have fibrillary appearance. Focal reduplication of the glomerular capillary basement membranes is also seen. A few intracapillary polymorphonuclear leukocytes are present. The specimen is from an adult patient without known etiology of HUS. (Periodic acid-Schiff reaction.) |
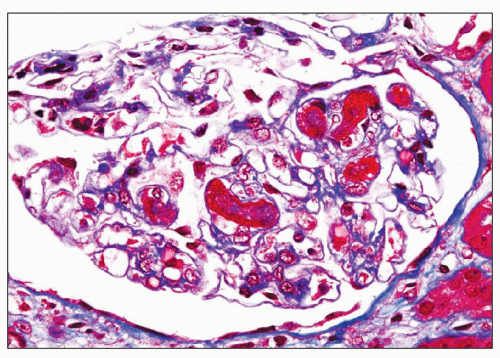 FIGURE 18.4 Thrombotic microangiopathy, associated with cyclosporine administration. Some of the glomerular capillary lumina are occluded by thrombi. No additional significant pathologic changes are present. (Masson trichrome.) |
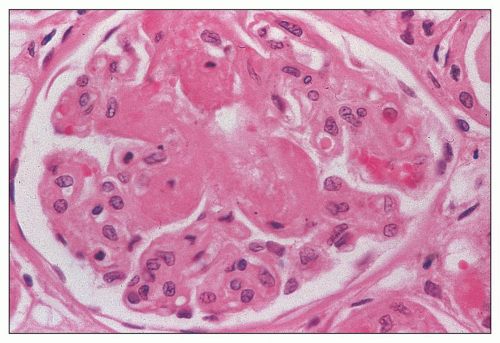 FIGURE 18.5 Hemolytic-uremic syndrome, atypical form. Some of the glomerular capillary tufts are permeated by eosinophilic acellular material. This change is often described as fibrinoid necrosis. Intraluminal thrombi are also present. (H&E) (From Kern WF, et al. Atlas of Renal Pathology. W.B. Saunders Company, 1999, with permission.) |
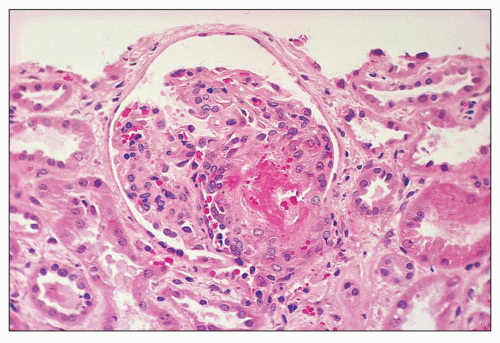 FIGURE 18.6 Thrombotic microangiopathy, secondary to abruptio placentae. The dilated vascular pole is occluded by a thrombus. The change is similar to that seen on Figure 18.10, but no significant chronicity with reduplication of the basement membranes is present. (H&E) |
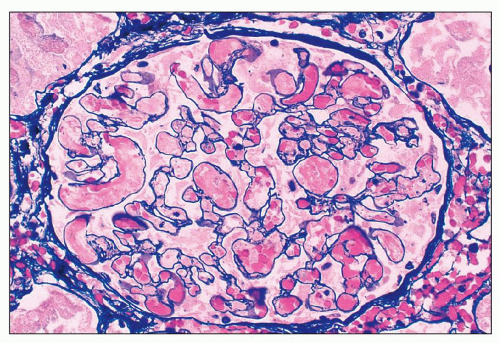 FIGURE 18.7 Thrombotic microangiopathy in a patient with primary antiphospholipid antibody syndrome. Some of the glomerular capillary lumina are occluded by fibrin thrombi; the rest of the capillaries are congested. Glomerular capillary congestion in HUS is often referred to as “glomerular paralysis.” Mesangiolysis is also apparent. (Methenamine-silver.) |
poorly because of mesangial edema. The borders of the dissolving mesangium are hazy, and the mesangial matrix is difficult to identify. No associated inflammatory reaction or fibrin deposition is usually seen (314). Eventually, the glomerular basement membranes become unanchored from the underlying dissolving mesangial mass, leading to markedly dilated, sometimes cystic capillaries (Fig. 18.9). A particularly severe and widespread form of mesangiolysis can occur in aHUS after bone marrow transplantation or mitomycin therapy. However, in classic HUS, mesangiolysis has rarely been described. Mesangiolysis can also be seen in diabetes mellitus, various forms of glomerulonephritis, and transplant glomerulopathy (315). “Healing” of mesangiolysis may lead to proliferating or sclerosing glomerular changes as the disease progresses. In the late stage of mesangiolysis, the mesangium may be thickened by pale fibrillary (sclerotic) material. This process of healing and sclerosing may lead to a distinctive pattern of glomerular sclerosis (“bland sclerosis”) characterized by loss of glomerular cells and capillary lumina but with at least partial preservation of the lobular architecture.
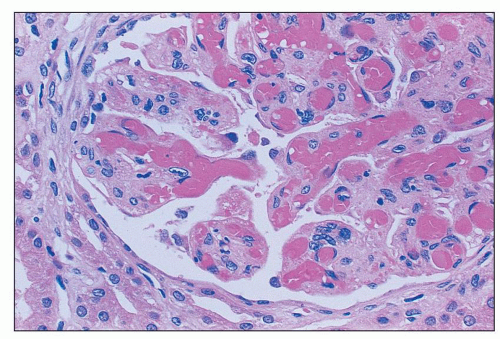 FIGURE 18.8 Thrombotic thrombocytopenic purpura. Most of the glomerular capillary lumina are occluded by homogenous eosinophilic thrombi. The extent of the glomerular capillary thrombosis in TTP is variable; however, it is often mild and patchy. The specimen is from a 40-year-old obese African American female who died a few days after initial presentation. Severe ADAMTS13 deficiency and a strong ADAMTS13 inhibitor were demonstrated in her serum. (H&E) |
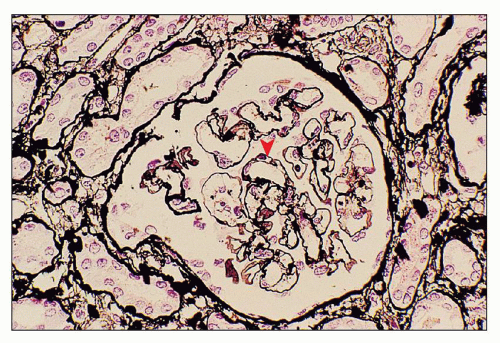 FIGURE 18.9 Thrombotic microangiopathy, associated with mitomycin administration. Ectatic glomerular capillary lumina are present as a result of mesangiolysis. Focal reduplication of the glomerular capillary basement membranes is also seen (arrowhead). (Methenamine-silver.) |
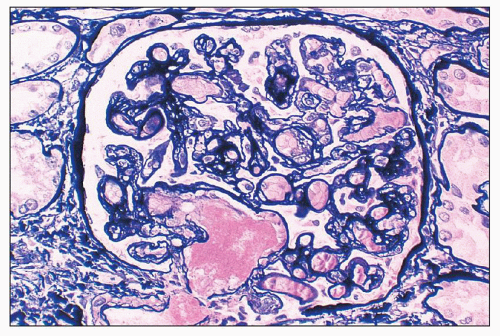 FIGURE 18.10 Thrombotic microangiopathy, postpartum. The dilated infundibulum is occluded by homogenous eosinophilic material (intraluminal thrombus). Extensive reduplication of the glomerular capillary basement membranes indicates developing chronicity. (Methenamine-silver.) |
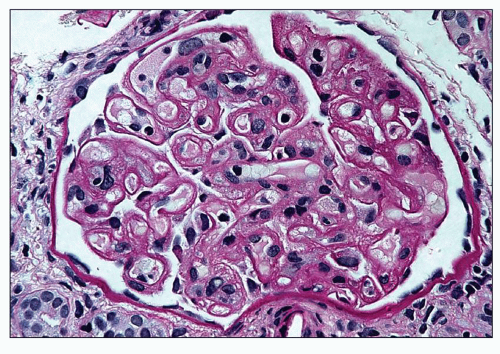 FIGURE 18.11 Thrombotic microangiopathy with extensive reduplication of the glomerular capillary basement membranes. This pattern is usually interpreted as chronic or advanced stage of TMA and may resemble a membranoproliferative pattern of glomerular injury. It should be emphasized that such extensive reduplication of the basement membranes as shown on the photograph is rarely seen. (Periodic acid-Schiff reaction.) |
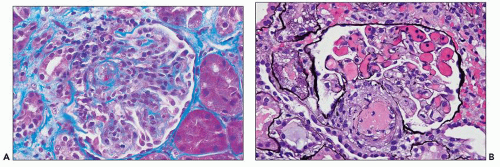 FIGURE 18.12 Thrombotic microangiopathy superimposed on lupus nephritis. A: The glomerulus is hypercellular with closure of some of the glomerular capillary lumina. There is a small thrombus in the arteriolar lumen. (Masson trichrome.) B: The arteriolar lumen is occluded by a thrombus. The glomerular capillary lumina are congested, but no capillary thrombi or cellular proliferation is present. Although this patient’s serum was positive for antiphospholipid antibodies, a similar picture can also be seen in patients who are antiphospholipid antibody negative. (Methenamine-silver.) |
necrosis tends to occur only at the hilum of the glomerulus, and it may only involve the thickened intima of the arterioles. More often, however, the media of the arteriole is also affected. Unlike in true leukocytoclastic vasculitis, acute inflammatory cell infiltrate is rarely seen in fibrinoid necrosis with TMA. As the disease progresses, the involved arterioles tend to become hyalinized, losing the staining reactions for fibrin. Hyalinized arterioles show homogenous eosinophilic, refractile, strongly PAS-positive acellular material accumulated in the intima or media. Fibrin thrombi may also be seen in the afferent arterioles, and these may continue into the glomerular capillary tuft (see Figs. 18.10 and 18.12). One of the most conspicuous features of TTP is the presence of eosinophilic, platelet-rich granular thrombi in terminal renal interlobular arteries, or more commonly, in afferent arterioles. The most common site is the junction of the afferent arteriole and the glomerular tuft, sometimes called the infundibulum (Fig. 18.14). The thrombotic material in the lumen may merge with the arteriolar wall; therefore, it is often difficult to distinguish between fibrinoid necrosis of the arteriolar wall and the fibrin thrombus in the lumen. Sometimes both fresh and organizing thrombotic lesions can be recognized. Aneurysmal dilatation of arterioles sometimes takes place, particularly in the hilar region of the glomerulus, in patients with TTP (4,316). In addition, the arterioles may show proliferation of cells assumed to be endothelial cells; this change was first reported by Baehr et al. (2) and was later confirmed by several authors. The cellular proliferation in the arterioles is sometimes a prominent feature, and the collections of cells, often concentrically arranged, may attain the size of glomeruli. Capillary channels with or without an edematous extracellular matrix can sometimes be recognized. Lipidcontaining macrophages may be present in the intima of small renal arteries undergoing proliferative changes. Because these proliferations resemble glomeruli, they are referred to as glomera, or glomeruloid structures (317). Glomeruloid structures are typically described in TTP but may also be seen in HUS, SLE, and various forms of glomerulonephritis (317).
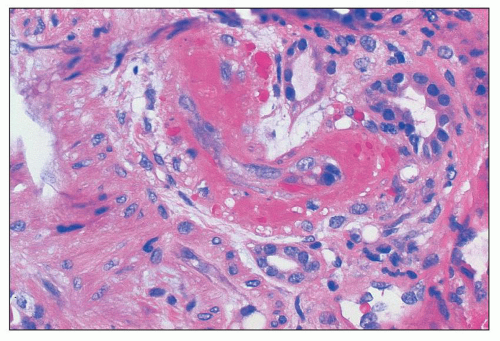 FIGURE 18.13 Thrombotic microangiopathy with arteriolar fibrinoid necrosis in a patient with systemic lupus erythematosus (SLE). In the setting of SLE, this finding is also referred to as lupus vasculopathy. Note the lack of inflammatory reaction in the vessel wall. Such finding can also be associated with intraluminal thrombosis affecting arterioles and/or glomerular capillary lumina. (H&E) |
cells and delicate connective tissue fibrils. The arterial changes may result in severe narrowing or occlusion of the vascular lumen with consequent reduction in blood flow. These severe vascular changes are associated with a poor prognosis (318) and are responsible for the secondary ischemic glomerular changes such as shrinkage of glomerular capillary tufts and wrinkling and thickening of the capillary walls. Fibrous replacement of the thickened intima is a later change in the interlobular arteries. Occasionally, thrombosis and recanalization can also be seen in the interlobular arteries (Fig. 18.17). Changes similar to those seen in the interlobular arteries can also be present, although less frequently, in the larger (arcuate and intralobar) arteries. Especially in older patients with preexisting chronic hypertension, the acute vascular lesions of TMA may be superimposed on chronic vascular changes such as intimal fibroplasia, medial hypertrophy, or arteriolar or arterial hyalinosis.
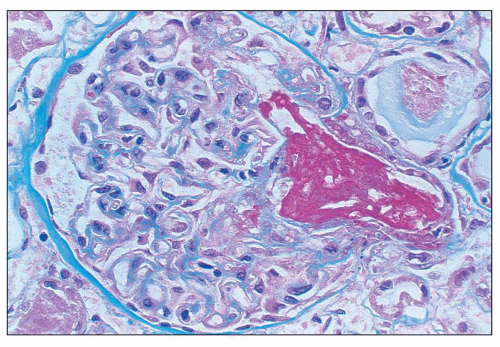 FIGURE 18.14 Thrombotic thrombocytopenic purpura. The infundibulum (i.e., vascular pole) is occluded by a large thrombus. The glomerular capillary walls are thickened; however, the glomerular capillary lumina are patent. (Masson trichrome.) |
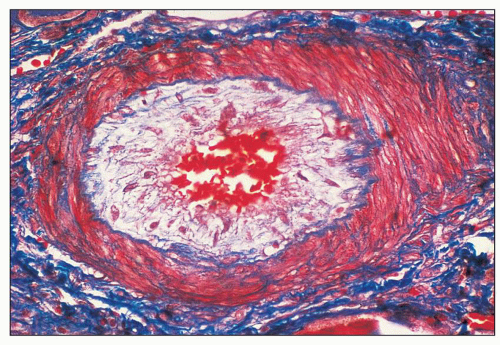 FIGURE 18.15 Thrombotic microangiopathy secondary to malignant hypertension. The small interlobular artery shows the edematous intima containing few myointimal cells (“mucoid intimal hyperplasia”). The patient presented with severe (“malignant”) hypertension and acute renal failure. (Lendrum stain.) |
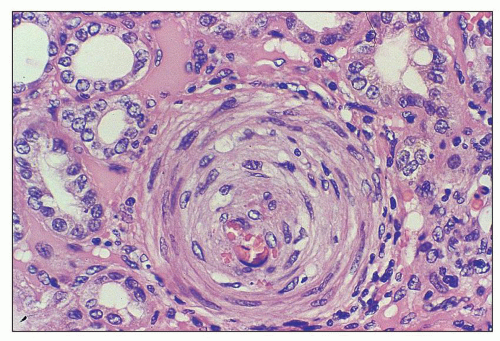 FIGURE 18.16 Hemolytic-uremic syndrome, atypical form. Prominent circumferential intimal cellular proliferation in a small interlobular artery in a nephrectomy specimen from a 5-year-old boy with aHUS. The child had severe hypertension. (H&E) |
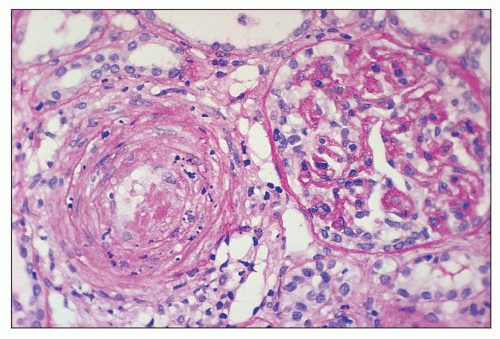 FIGURE 18.17 Hemolytic-uremic syndrome, atypical form. The interlobular artery shows luminal thrombus with nuclear debris in the arterial wall. The glomerulus exhibits ischemic features with thickening and wrinkling of the glomerular capillary basement membranes. The specimen is from an adult patient without known etiology of HUS. (Periodic acid-Schiff reaction.) |
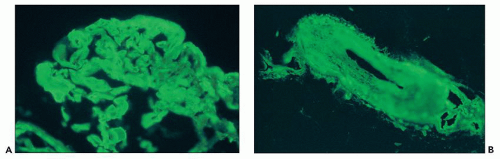 FIGURE 18.18 Hemolytic-uremic syndrome (TMA). The glomerular capillary walls and lumina (A) and the arterial wall (B) show strong fibrinogen positivity. The patient presented with severe (“malignant”) hypertension, acute renal failure, MAHA, and thrombocytopenia. (Direct immunofluorescence.) |
or fibrin and weak factor VIII-related antigen staining was observed. Electron microscopic analysis showed numerous platelets in the glomerular capillary thrombi in TTP. The authors concluded that thrombi in TTP are composed of platelets. The strong subendothelial factor VIII-related antigen positivity was interpreted by these investigators as suggesting that the hyaline deposits are not a result of increased vascular permeability, but rather platelet thrombi incorporated into the vascular wall. Similarly, a more recent autopsy study of 25 patients with TTP and 31 patients with HUS demonstrated the histochemical and immunohistochemical differences in thrombi (320). HUS lesions contained a large component of fibrin, highlighted by phosphotungstic acid-hematoxylin stain, but only a few platelets or vWF (320,321). On the other hand, TTP-associated arterial thrombi had abundant platelets that stained for antibody to factor VIII. The TTP microthrombi were also rich in vWF but contained very little fibrin (322). Despite these apparent differences in HUS and TTP microthrombi, given the extensive clinical overlap between aHUS and TTP, the histologic accuracy and validity of these findings are unclear.
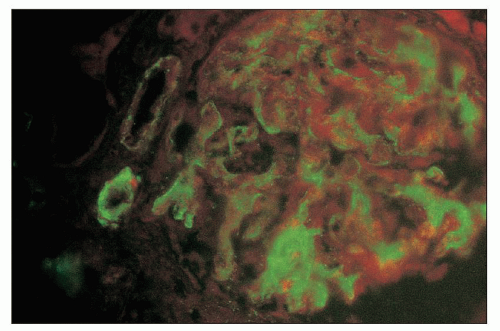 FIGURE 18.19 Thrombotic microangiopathy. IgM glomerular positivity in a patient with postpartum HUS. The arteriolar wall is also positive. (Direct immunofluorescence.) |
glomerulonephritis may be absent or reveal aberrant features. The relatively high incidence of hepatitis C virus infection in association with transplant glomerulopathy pattern glomerular injury posttransplantation (254) is suggestive of a potentially significant etiologic role of hepatitis C in this process. The significance of hepatitis C infection in causing chronic TMA-like changes in an allograft may have been underestimated until recently (325,326).
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Renal Disease Caused by Hypertension
Renal Disease Caused by Hypertension
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


