pericarditis, acute nephritis, and hemorrhage from the mucous surface.” In later writings, he emphasized the arthritis, pulmonary, and central nervous system manifestations while remarking that the arthritis was typically nondeforming. He observed the variable clinical presentation and remitting and relapsing nature of the condition: “Recurrence is a special feature of the disease and attacks may come on month after month or even throughout a long period of years… . The attacks may not be characterized by skin manifestations; the visceral symptoms alone may be present, and to the outward view, the patient may have no indications whatever of erythema exudativum” (5).
(Table 14.2). It defined six major classes and a large number of subclasses, with emphasis on distribution, activity, and chronicity of the lesions. Although much more detailed and precise, this revised classification was not as widely accepted because of its cumbersome reliance on subclasses. A third classification was proposed in 2003 by a large consensus conference of renal pathologists, nephrologists, and rheumatologists that was organized jointly by the International Society of Nephrology (ISN) and the Renal Pathology Society (RPS) (Table 14.3). This new classification, which has been entitled “ISN/RPS WHO classification revisited,” retains the simplicity of the original 1974 WHO classification while incorporating some of the refinements introduced by the 1982 revised WHO classification (21,22). The ISN/RPS system provides more detailed and more precise pathologic criteria for each class, which facilitates greater reproducibility among pathologists. This updated consensus classification is widely accepted and has superseded prior classifications.
TABLE 14.1 Original (1974) WHO classification of lupus nephritis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
TABLE 14.2 Modified (1982) WHO classification of lupus nephritis | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
diagnosed patient with SLE who lacks clinically apparent renal disease. However, because some cases of severe lupus nephritis are clinically “silent,” this liberal approach has some proponents (26). Most nephrologists and rheumatologists would agree that the new appearance of any significant marker of renal disease such as hematuria, proteinuria, nephrotic syndrome, or renal insufficiency at any time in the course of SLE is ample justification for renal biopsy. The recent ACR guidelines for management of lupus nephritis advocate that renal biopsy should be performed in any patient with clinical
evidence of renal involvement defined as one of the following: (a) increasing serum creatinine without compelling alternative causes (such as sepsis, hypovolemia, or medication); (b) confirmed proteinuria of ≥1.0 g per 24 hours or by spot urine protein:creatinine ratio; or (c) combinations of proteinuria ≥0.5 g plus hematuria defined as ≥5 RBCs per hpf or proteinuria ≥0.5 g plus cellular casts (27). Moreover, lupus nephritis is one of the few renal diseases for which “follow-up” renal biopsies are routinely performed in some centers 6 months or more after therapy to gauge the efficacy of treatment and guide further therapeutic management. Repeat renal biopsies are also frequently performed in any patient with a sudden change in renal findings (e.g., new onset of proteinuria, new activity of the urinary sediment, declining glomerular filtration rate [GFR]) that may presage a transformation in the class of lupus nephritis, or reactivation of disease requiring reinstitution or adjustment of therapy.
TABLE 14.3 ISN/RPS classification of lupus nephritis (LN) (2004) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
TABLE 14.4 Integration of LM, IF, and EM findings by the WHO class | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
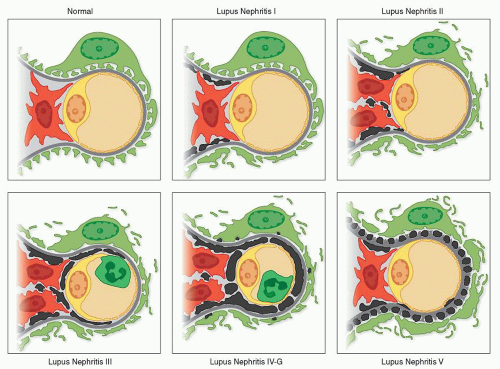 FIGURE 14.1 Normal capillary with intact podocyte foot processes and no electron-dense deposits. Class I lupus nephritis with minimal mesangial deposits, focal foot process effacement, and no mesangial hypercellularity. Class II lupus nephritis with substantial mesangial deposits, mesangial hyperplasia, and more extensive foot process effacement. Class III lupus nephritis with scanty subendothelial deposits, mesangial hyperplasia, endocapillary leukocytes, and extensive foot process effacement. Class IV-G lupus nephritis with numerous subendothelial and a few subepithelial deposits, mesangial hyperplasia, endocapillary leukocytes, and extensive foot process effacement. Class V lupus nephritis with numerous subepithelial and a few subendothelial and mesangial deposits and extensive foot process effacement. |
general remarks serve as a preface to the more detailed consideration of the WHO classification that follows. Many of these morphologic features pertain to more than one WHO class of lupus nephritis.
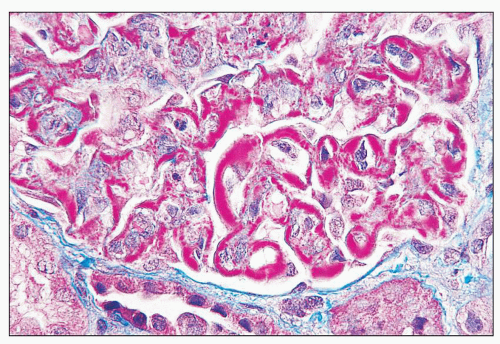 FIGURE 14.2 Lupus nephritis class IV. Trichrome stain highlights the presence of global subendothelial fuchsinophilic deposits. (Masson trichrome, × 500.) |
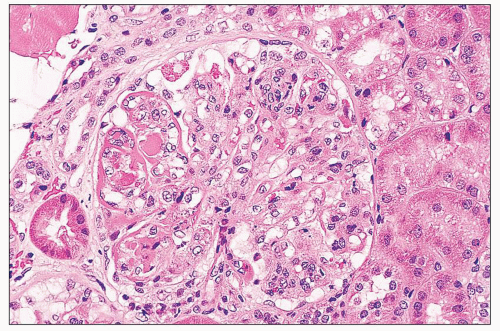 FIGURE 14.3 Lupus nephritis class IV. Glomerular capillary walls are segmentally thickened by wire-loop deposits. An intraluminal deposit forms a “hyaline thrombus” in one capillary, and there is global endocapillary proliferation. (H&E; × 320.) |
contour, similar to that observed in membranoproliferative glomerulonephritis. Not surprisingly, mesangial extension or “interposition” frequently accompanies this finding.
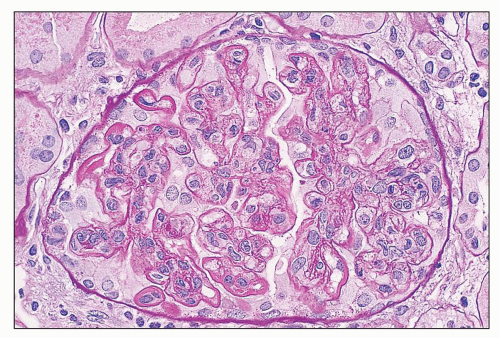 FIGURE 14.4 Lupus nephritis class IV. PAS stain highlights the thickening of the glomerular capillary walls by numerous subendothelial deposits. (PAS; × 500.) |
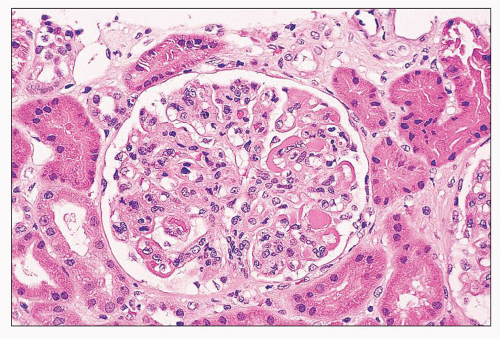 FIGURE 14.5 Lupus nephritis class IV. In addition to wire-loop deposits, there are segmental intraluminal deposits forming “hyaline thrombi.” (H&E; × 320.) |
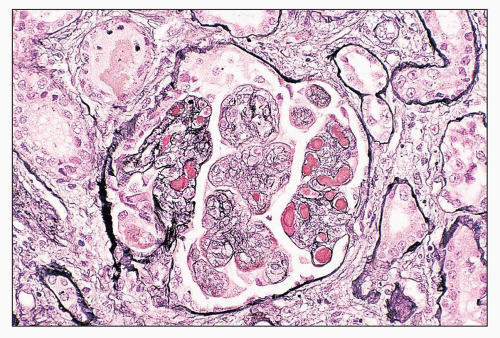 FIGURE 14.6 Lupus nephritis class IV. Silver stain is useful to highlight the intraluminal location of the glomerular capillary fibrin thrombi. Some GBMs appear double contoured. (Jones methenamine silver, × 320.) |
by staining for fibrin-related antigens by immunofluorescence. Of these two histochemical stains, PTAH is less reliable, because it sometimes stains immune deposits and fibrin, whereas the Lendrum stain is the more sensitive and specific for fibrin. Of considerable help in differentiating true fibrin thrombi is the appearance of the material in H&E-stained sections. Fibrin often has a darkly eosinophilic fibrillar appearance, whereas hyaline thrombi of the immune deposit type are more lightly eosinophilic, with a homogeneous, glassy, smooth texture.
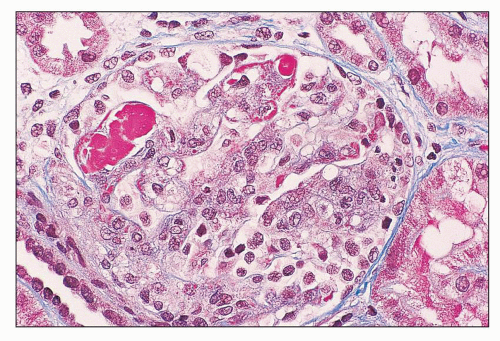 FIGURE 14.7 Lupus nephritis class IV. The hyaline thrombi stain red against the blue-staining glomerular capillary walls. (Masson trichrome, × 500.) |
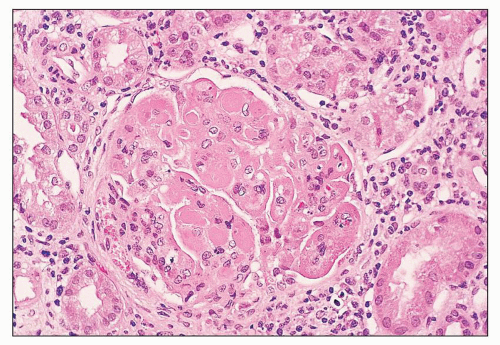 FIGURE 14.8 Lupus nephritis class IV. Extensive intraluminal and subendothelial deposits form “hyaline thrombi” and wire loops. Endocapillary proliferation is less conspicuous in the capillaries with voluminous deposits. (H&E; × 320.) |
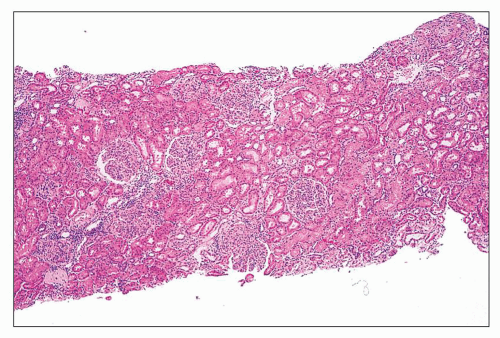 FIGURE 14.9 Lupus nephritis class IV. There is relatively uniform diffuse and global endocapillary proliferation. (H&E; × 100.) |
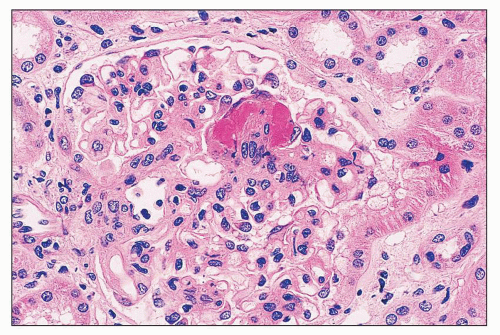 FIGURE 14.10 Lupus nephritis class III. There is segmental fibrinoid necrosis with neutrophil infiltration and pyknosis. (H&E; × 500.) |
(35,36,37) define cellular crescents as “aggregates comprising two or more layers of proliferating visceral and parietal epithelial cells with infiltrating mononuclear cells lining one fourth or more of the interior circumference of the Bowman capsule.” This reasonable definition allows differentiation of crescents from the single layer of reactive hyperplastic visceral epithelial cells commonly encountered in glomeruli with membranous features or undergoing sclerosis. Cellular crescents of inflammatory origin must be differentiated from the epithelial cell proliferation that accompanies the collapsing variant of focal segmental glomerulosclerosis (FSGS) (collapsing glomerulopathy), which has been described in some patients with SLE (see Lupus Podocytopathy).
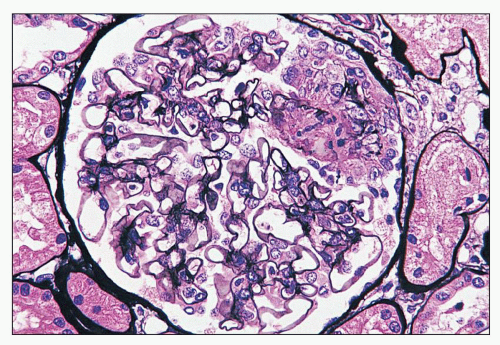 FIGURE 14.11 Lupus nephritis class III. The segmental necrotizing lesion displays segmental rupture of the GBM. (Jones methenamine silver, × 500.) |
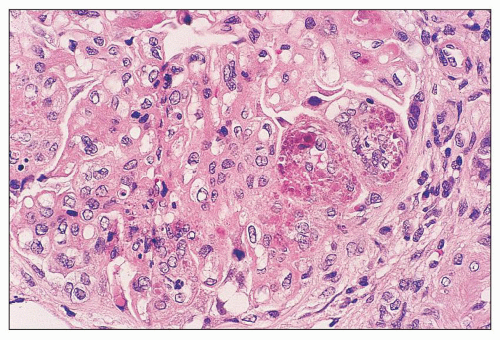 FIGURE 14.12 Lupus nephritis class IV. Several glomerular capillaries contain hematoxylin bodies consisting of smudged, coarsely clumped basophilic material. Another lobule contains karyorrhectic nuclear debris. (H&E; × 500.) |
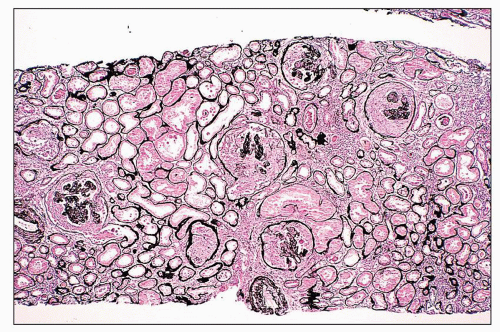 FIGURE 14.13 Lupus nephritis class IV. A severe example with extensive cellular crescents. Although the glomerular tufts are compressed, endocapillary proliferation is evident. (Jones methenamine silver; × 80.) |
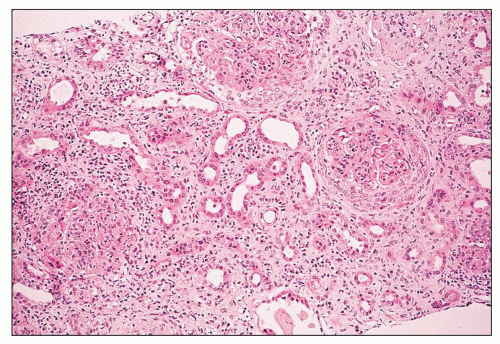 FIGURE 14.14 Lupus nephritis class IV. Initial renal biopsy shows diffuse endocapillary proliferation with focal crescents and severe interstitial inflammation. (H&E; × 80.) |
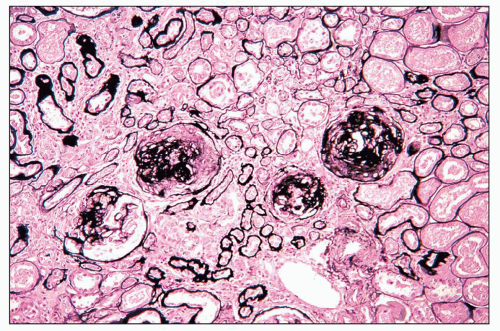 FIGURE 14.15 Lupus nephritis class IV. Despite aggressive therapy, repeat renal biopsy 2 years later shows progression to segmental and global glomerulosclerosis with focal fibrous crescents. There is marked reduction in the degree of interstitial inflammation. (Jones methenamine silver, × 80.) |
expanded and organized into aggregates with focal germinal centers containing follicular dendritic cells (51). The B cells have an immunoglobulin (Ig) repertoire consistent with somatic hypermutation and organ-intrinsic adaptive immune responses. The close proximity of B and T cells suggests that in addition to in situ antibody secretion, resident B cells are contributing to local inflammation by presenting major histocompatibility complex (MHC) class II-restricted antigens to T cells (51). Intrarenal production of anti-dsDNA antibody by resident plasma cells has also been described in the New Zealand Black (NZB)/W murine model of lupus nephritis, indicating that the kidney can be a site of autoantibody formation (52). IL-17-producing double-negative (CD4–/CD8–) T cells have also been identified in the renal interstitium (53). Several studies using immunostaining for macrophages confirmed the large number of interstitial and intratubular macrophages in diffuse proliferative lupus nephritis and found good correlations between tubular macrophages and serum creatinine, proteinuria, and progression to renal insufficiency (34,54). Urinary biomarkers that reflect the severity and activity of interstitial inflammation in human lupus biopsies include monocyte chemotactic protein 1, hepcidin (produced by monocytes in response to INFα), and liver-type fatty acid-binding protein (produced by the proximal tubule in response to injury) (55).
By immunofluorescence, the deposits have a granular to semilinear texture that has a tendency to outline the profiles of tubular basement membranes and interstitial capillaries or to form punctate aggregates in the interstitial collagen (Fig. 14.18). Interstitial capillary immune deposits are particularly specific for lupus nephritis (Fig. 14.19).
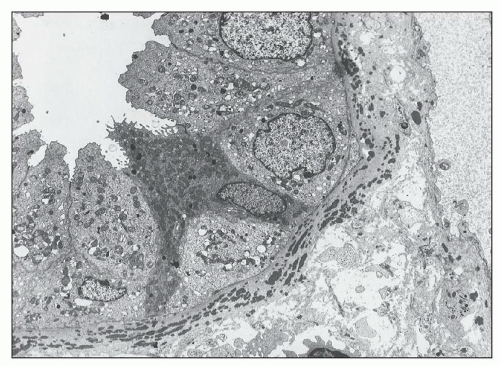 FIGURE 14.16 Lupus nephritis class IV. The electron micrograph shows electron-dense deposits within a tubular basement membrane and, to a lesser extent, in the adjacent interstitial collagen. (× 2000.) |
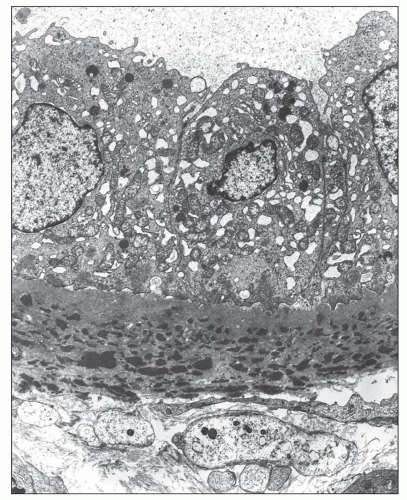 FIGURE 14.17 Lupus nephritis class IV. High-power view shows a lamellated network of tubular basement membrane splayed around the tubular basement membrane deposits. (Electron micrograph, × 4000.) |
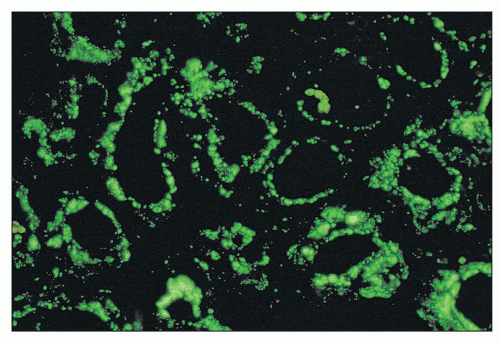 FIGURE 14.18 Lupus nephritis class IV. The immunofluorescence micrograph shows abundant granular deposits of IgG within the tubular basement membranes and interstitium. (× 200.) |
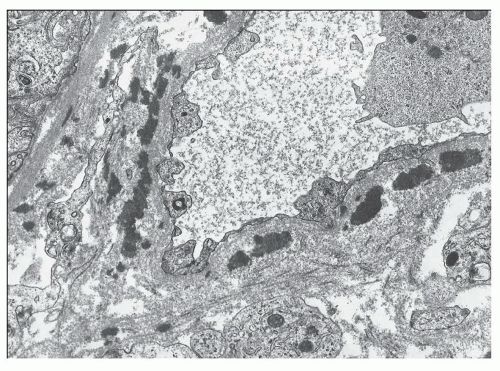 FIGURE 14.19 Lupus nephritis class IV. This electron micrographs shows numerous electron-dense deposits within the wall of an interstitial capillary. (× 6000.) |
thrombotic vasculopathy, true inflammatory vasculitis, and nonspecific arteriosclerosis (Table 14.5). Because assessment of vascular lesions is not factored into the ISN/RPS classification of lupus nephritis or formulation of the activity and chronicity index according to the National Institutes of Health (NIH) criteria, renal vascular lesions run the risk of being overlooked. This is especially the case when vascular lesions are focally distributed. To accurately classify renal vascular lesions, vessels must be systematically examined by light, immunofluorescence, and electron microscopy. The morphologic findings must then be analyzed in the context of particular clinical syndromes that may complicate SLE, including APL antibody syndrome (alternatively known as anticardiolipin syndrome), thrombotic thrombocytopenic purpura, renal vein thrombosis (RVT), and accelerated hypertension.
TABLE 14.5 Vascular lesions in lupus nephritis | ||
|---|---|---|
|
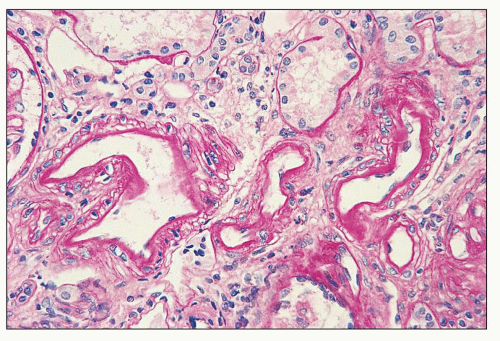 FIGURE 14.20 Lupus nephritis class IV. Intimal immune deposits in the walls of interlobular arteries have caused slight thickening of the intimal basement membrane without significant luminal narrowing. (PAS; × 320.) |
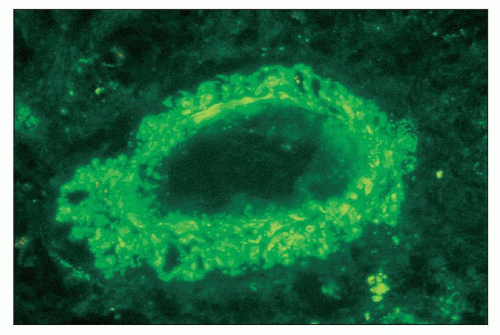 FIGURE 14.21 Lupus nephritis class IV. The fluorescence micrograph shows abundant granular staining for IgG within the intima and media of an interlobular artery. (× 400.) |
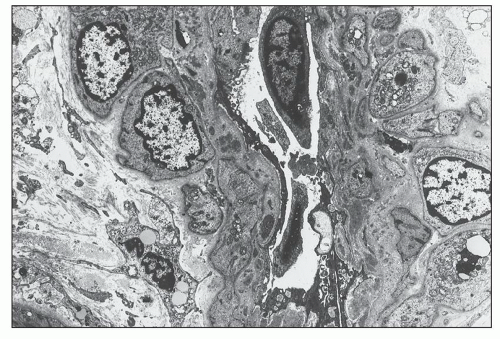 FIGURE 14.22 Lupus nephritis class IV. The electron micrograph shows small, granular, electron-dense deposits within the subendothelial basement membrane of an interlobular artery. (× 2875.) |
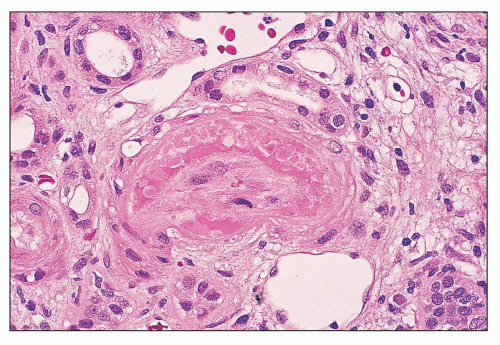 FIGURE 14.23 Lupus vasculopathy. The lumen of an arteriole is severely narrowed by intimal deposits of intensely eosinophilic material suggestive of fibrin admixed with more lightly eosinophilic immune deposits. The endothelium is necrotic; there is no inflammation of the vessel wall. (H&E; × 500.) |
(Fig. 14.26). By light microscopy, the affected vessels are occasionally narrowed or occluded by intraluminal fibrin and platelet thrombi, which may be associated with endothelial swelling and denudation (Fig. 14.27). The products of coagulation may penetrate the intima and sometimes contain fragmented erythrocytes. There is no overt leukocyte infiltration of the media, although a rare neutrophil or lymphocyte may be entrapped in the luminal thrombosis. In the interlobular arteries, the intima may have a more mucoid, edematous appearance, with “onion skin” myointimal proliferation. By fluorescence microscopy, the affected vessels usually reveal intense, dominant staining for fibrin-related antigens, with variable positivity for IgM and C3. In contrast to lupus vasculopathy, IgG is usually absent from these lesions. Recently, C4d has been identified as a potential marker of glomerular thrombotic microangiopathy in lupus nephritis, irrespective of whether there was an associated APL antibody (94). The incidence of thrombotic lesions resembling HUS/TTP was 8% of lupus nephritis patients according to a collaborative Italian study (84). Their presence was associated with reduced (66.3%) 5-year renal survival compared with 89.6% for lupus nephritis patients without renal vascular lesions. They may be superimposed on virtually any class of lupus nephritis including classes II (95), III (95), and IV (96).
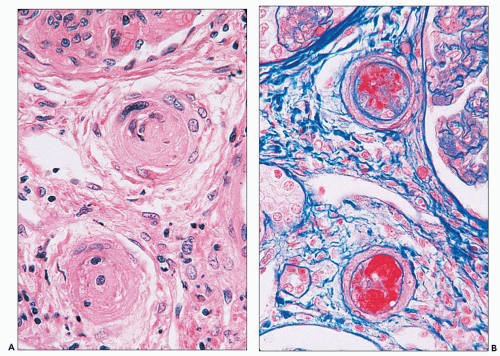 FIGURE 14.24 Lupus vasculopathy. A double panel shows occlusion of preglomerular arterioles by eosinophilic deposits (A) that stain positive with Lendrum stain for fibrin (B). (× 500.) |
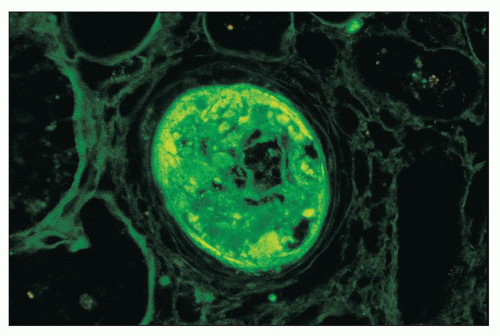 FIGURE 14.25 Lupus vasculopathy. An interlobular artery shows staining for IgA within its expanded intima. Similar deposits were seen for IgG, IgM, C3, C1q, and fibrinogen. The vessel lumen is nearly occluded. (× 600.) |
skin lesions, abdominal pain and bleeding, and neurologic symptoms, including reduced consciousness, seizures, transient pareses, and coma (95,98,99). Most well-documented cases have evidence of thrombocytopenia and microangiopathic hemolytic anemia. The results of other coagulation studies (e.g., prothrombin time, partial thromboplastin time, fibrin degradation products) have been normal, and the lupus anticoagulant is absent in most patients (83,95). Some cases have been linked to autoantibody to the von Willebrand factor cleaving protease ADAMTS-13 (100,101). An association with decreased serum complement factor H levels in some individuals suggests a role for dysregulation of the alternative complement pathway (101,102). Renal manifestations of the TTP-like process range from asymptomatic urinary abnormalities to mild reduction in renal function to fulminant oligoanuric renal failure requiring dialysis. Hypertension has been absent in most cases (95,97,103) but present in others (84). The TTP has responded to the same therapeutic strategies used in idiopathic TTP. In the modern era, plasma exchange and plasma infusion have achieved greatly improved survival compared with older forms of therapy, including steroids, antiplatelet agents, and splenectomy.
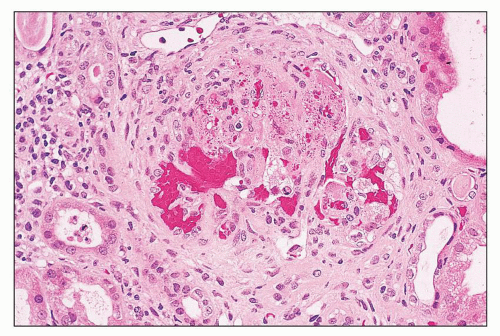 FIGURE 14.26 Thrombotic thrombocytopenic purpura-like syndromein SLE. This glomerulus displays features of acute thrombotic microangiopathy, including marked glomerular capillary congestion, endothelial swelling and necrosis, and glomerular capillary thrombosis with entrapment of fragmented erythrocytes (i.e., schistocytes). (H&E; × 320.) |
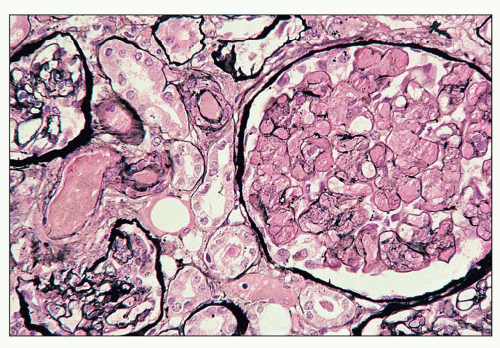 FIGURE 14.27 Thrombotic thrombocytopenic purpura-like syndrome in SLE. The preglomerular arteriole and many glomerular capillaries are occluded by fibrin thrombi. The arteriolar and glomerular endothelium appears denuded. (Jones methenamine silver, × 320.) |
a syndrome of pregnancy-associated thrombotic microangiopathy in 12 young women with APL syndrome, 8 of whom had no evidence of SLE and 4 of whom had SLE, including 2 with proliferative glomerulonephritis (135). D’Agati et al. (129) stressed that the occurrence of thrombotic microangiopathy in APL syndrome could not be accounted for by secondary intravascular coagulation caused by active proliferative and necrotizing lupus nephritis.
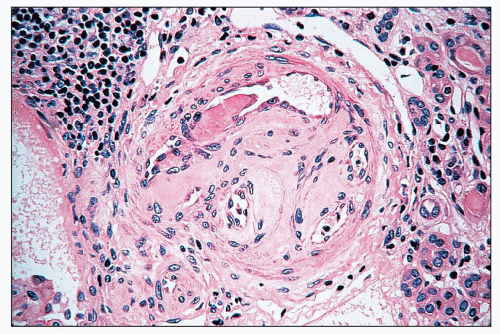 FIGURE 14.28 Lupus anticoagulant syndrome in SLE. An interlobular arteries contains fresh and recanalized thrombus. (H&E, × 400.) |
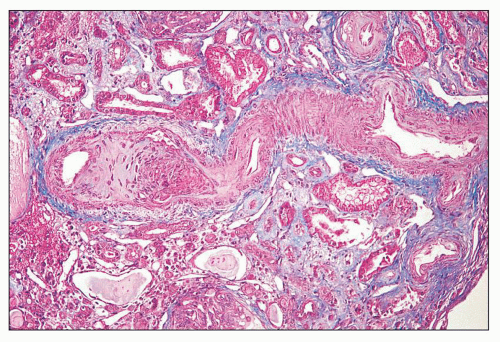 FIGURE 14.29 Lupus anticoagulant syndrome in SLE. An interlobular artery is narrowed by organizing thrombus. There is adjacent tubular atrophy and interstitial fibrosis. (Masson trichrome; × 200.) |
closely linked to APL syndrome and may be a manifestation of drug-induced APL antibody, particularly from procainamide. IgA APL antibody has been linked to a higher incidence of thrombocytopenia in some reports. Many patients have APL antibodies of more than one class. False-positive Venereal Disease Research Laboratory (VDRL) test results have been obtained from APL antibodies reactive with the cardiolipin and phosphatidylcholine component of beef heart used in this assay.
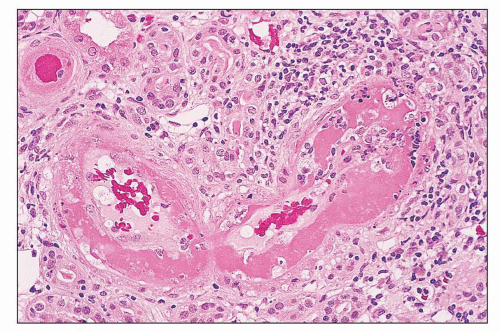 FIGURE 14.30 Acute necrotizing vasculitis in SLE. This patient with class IV lupus nephritis and multisystem vasculitis had necrotizing, inflammatory vasculitis of several small interlobular arteries. There is transmural infiltration by neutrophils and lymphocytes, with abundant intimal fibrin and necrosis of the media. (H&E; × 320.) |
and C4 attests to the activation of complement by the classic pathway (153). The staining pattern is called “full house” (meaning 3 of a kind and 2 of a kind in poker parlance) when deposits containing all three immunoglobulin classes (IgG, IgM, and IgA) and both complement components (C3 and C1q) are present.
TABLE 14.6 Incidence of positive glomerular immunofluorescence for different antisera from four series | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
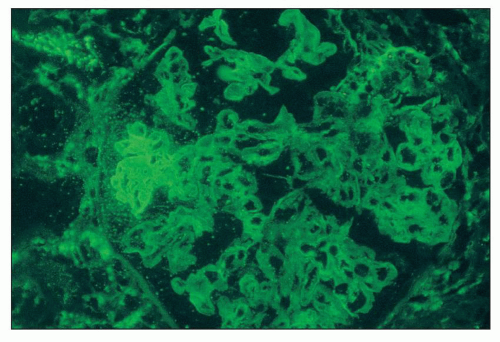 FIGURE 14.31 Lupus nephritis class III. There is strong staining for fibrin/fibrinogen in the distribution of a segmental necrotizing lesion. (Immunofluorescence micrograph, × 500.) |
rabbit antihuman IgG in a standard fashion. However, experimental data on murine lupus suggest that ANA may penetrate and bind to the nuclei of intact cells and therefore may occur in vivo (159,160). In cases with strong tissue ANA (particularly speckled type), the diffuse nuclear staining for IgG may interfere with detection of true immune deposits in glomeruli and the tubulointerstitium, requiring careful integration with the results of immunofluorescence staining for the other immune reactants.
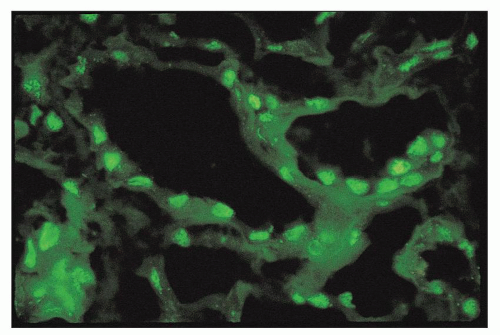 FIGURE 14.32 Lupus nephritis class IV. The immunofluorescence micrograph shows tubular ANA reactivity in this cryostat section stained for IgG. This finding has been referred to as “tissue ANA.” (× 600.) |
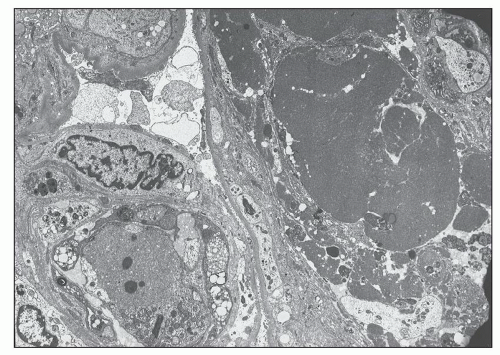 FIGURE 14.33 Lupus nephritis class IV. Electron micrograph shows luminal obliteration by a massive glomerular capillary electron-dense deposit corresponding to a “hyaline thrombus.” (× 2300.) |
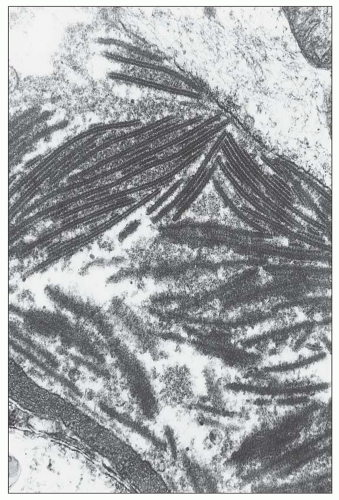 FIGURE 14.34 Lupus nephritis class IV. Electron micrograph shows an organized mesangial electron-dense deposit with tubulofibrillar substructure resembling that seen in cryoglobulinemia. (× 80,000.) |
with a mixed IgG-IgA-IgM cryoglobulin exhibited the same fingerprint substructures on ultrastructural study of the cryoprecipitate and the glomerular deposits (164,165). Moreover, fingerprint substructures similar to those in SLE have also been described in some cases of monoclonal IgG cryoglobulinemia (166). It appears that the fingerprint deposits are not specific for SLE but reflect the composition of the associated immune deposits.
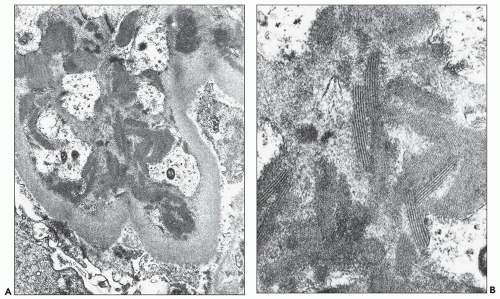 FIGURE 14.35 Lupus nephritis class IV. A large subendothelial deposit displays an organized substructure composed of parallel linear arrays resembling those seen in some forms of cryoglobulinemia. (Electron micrographs; A, × 10,000; B, × 30,000.) |
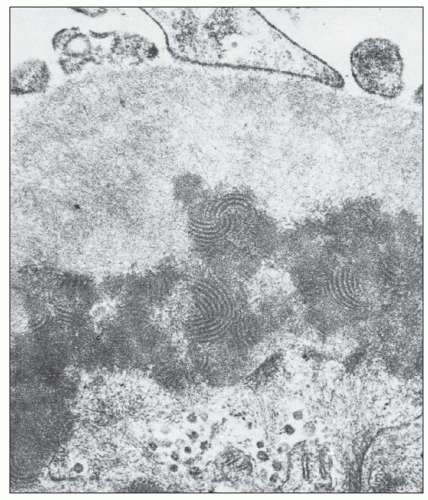 FIGURE 14.36 A subendothelial electron-dense deposit displays a fingerprint-whorled, lamellated substructure. (× 40,000.) (From Churg J, Grishman E. Ultrastructure of immune deposits in renal glomeruli. Ann Intern Med 1972;76:479.) |
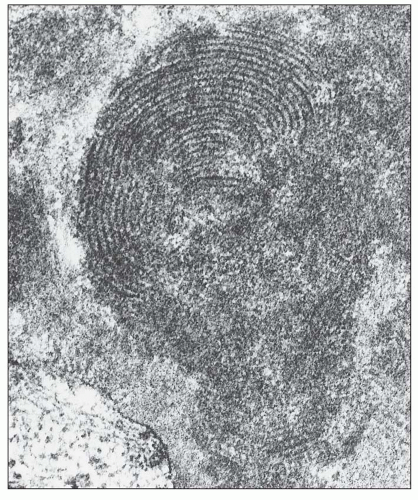 FIGURE 14.37 Electron micrograph of a subendothelial deposit with a fingerprint substructure shows it to be composed of curvilinear, concentric, membranous structures with fine cross-hatching. The remainder of the deposit has a granular texture. (× 100,000.) |
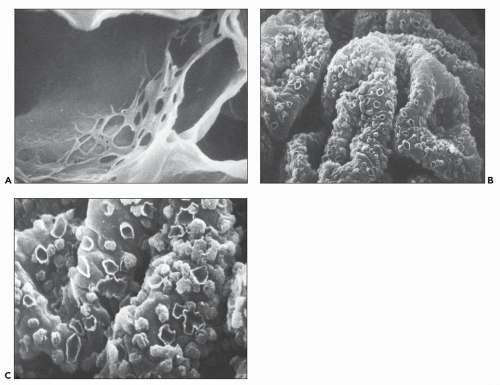 FIGURE 14.38 A: SEM from a patient with the membranoproliferative variant of lupus nephritis. Notice the web-like network on the inner aspect of capillary basement membrane, which probably represents subendothelial extension of mesangial matrix. (× 5500.) B: SEM of a glomerulus from a patient with membranous lupus nephritis shows discrete crater-like deformities, some containing “balls” of immune complex material. (× 687.) C: Higher-power view of the glomerulus in (B) shows crater-like deformities and immune complex-like balls in greater detail. (× 5500.) (From Weidner N, Lorentz WB. Scanning electron microscopy of the acellular glomerular and tubular basement membrane in lupus nephritis. Am J Clin Pathol 1986;85:135.) |
component is partly or completely enveloped by membranes. The second component, presumed to be cytoplasmic in origin, consists of aggregates of vesicles, vacuoles, glycogen granules, and other spheroidal or rod-shaped granules, some of which exhibit swollen cristae identifiable as degenerating mitochondria. In some cases, the characteristics of the granules are identical to those of specific granules of neutrophils. These cytoplasmic components are surrounded by a continuous or fragmented membrane, which probably represents the original plasma membrane. These structures are enclosed within large phagocytic vacuoles of cells that appear to be mesangial or monocytic in origin. Adjacent mesangial deposits are often identified surrounding the phagocytic mesangial cell. The morphologic appearance of these structures is similar to the description of the LE cell by Maldonado et al. (170). Grishman and Churg (171) published electron microscopic studies of hematoxylin bodies that occurred in arterial walls in the absence of inflammation.
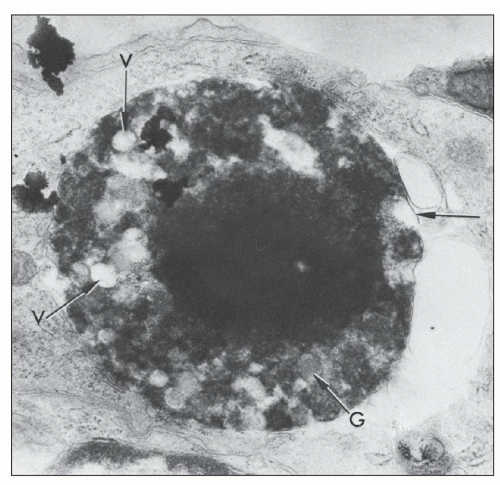 FIGURE 14.39 Lupus nephritis class IV. Electron micrograph of a hematoxylin body with a densely rounded central core of altered nuclear material and surrounding cytoplasmic elements consisting of degenerating organelles, all contained within a discontinuous membrane (arrows). V, vacuoles; G, granules. (× 21,402.) (From Cohen AH, Zamboni L. Ultrastructural appearance and morphogenesis of renal glomerular hematoxylin bodies. Am J Pathol 1977;89:105.) |
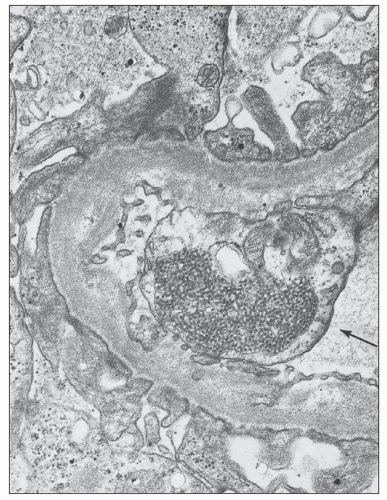 FIGURE 14.40 The electron micrograph shows TRIs within the endothelial cell (arrow) of a glomerular capillary and scattered, subepithelial, electron-dense deposits. (× 14,000.) (Courtesy of Dr. Antonovych, Armed Forces Institute of Pathology, Washington, DC.) |
alterations that are common to many other glomerular diseases manifesting altered glomerular permeability. These ultrastructural alterations include variable foot process effacement, condensation of cytoskeletal microfilaments, microvillous transformation, and cellular hypertrophy with increased organelles, including endoplasmic reticulum, mitochondria, and membrane bound vesicles, some of which contain electron-dense material suggestive of resorbed proteins.
TABLE 14.7 Frequencies of histologic patterns of lupus nephritis (ISN/RPS classification) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
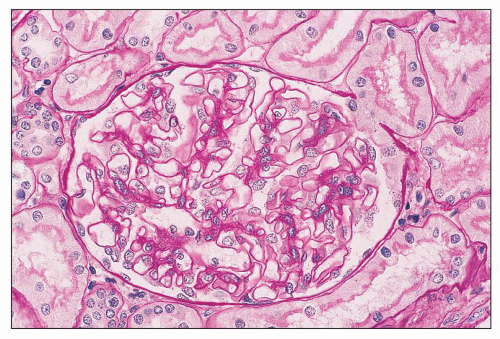 FIGURE 14.41 Lupus nephritis class I. The glomerulus is normal in cellularity, and the GBMs are unremarkable. (PAS; × 500.) |
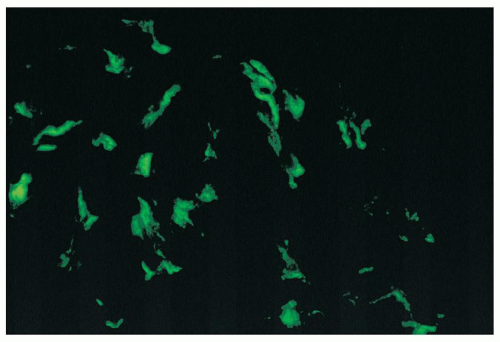 FIGURE 14.42 Lupus nephritis class I. Immunofluorescence microscopy shows delicate mesangial positivity for immunoglobulin G, although no abnormalities were seen by light microscopy. (× 600.) |
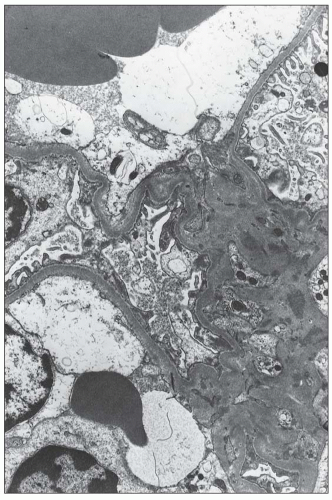 FIGURE 14.43 Lupus nephritis class I. The electron micrograph shows small, mesangial, electron-dense deposits. No deposits involve the peripheral GBMs, and foot processes are intact. (× 2500.) |
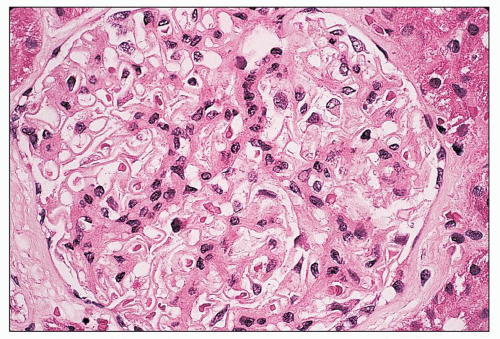 FIGURE 14.44 Lupus nephritis class II. There is mild, global, mesangial hypercellularity with thin capillary loops. (H&E, × 500.) |
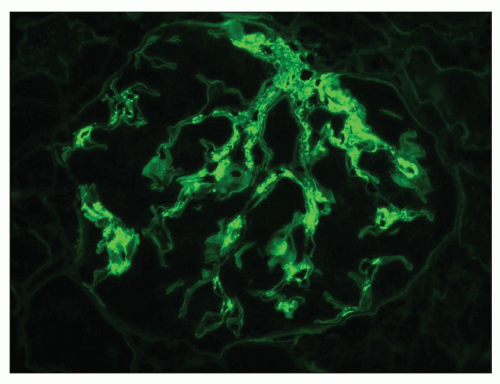 FIGURE 14.45 Lupus nephritis class II. Immunofluorescence microscopy shows deposits of IgG confined to the mesangium. (× 500.) |
problematic and were not adequately addressed by the original WHO classification system. According to the ISN/RPS classification, cases of mesangial proliferative lupus nephritis with rare minute subendothelial or subepithelial deposits (visible only by IF and/or EM) should be classified as class II (21,22). Most practicing renal pathologists deal with these atypical features by indicating in a note that the rare small subendothelial or subepithelial deposits observed in a minority of capillaries suggest the possibility that the glomerulonephritis may evolve into focal proliferative or membranous glomerulonephritis in the near future and that the patient should be carefully monitored. However, the presence of any sizeable (visible by LM) or numerous subendothelial or subepithelial deposits in an otherwise mesangial proliferative glomerulonephritis exceeds what is acceptable in pure class II disease and warrants a designation of class III, IV, or V depending on their distribution and quantity.
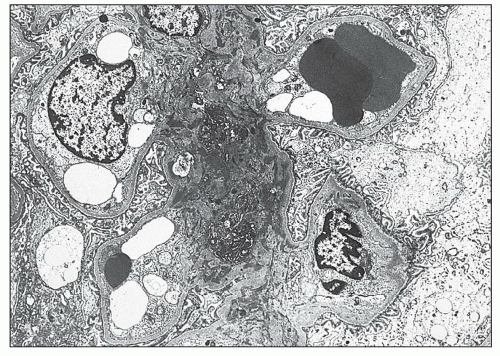 FIGURE 14.46 Lupus nephritis class II. The electron micrograph shows mesangial electron-dense deposits accompanied by mild mesangial hypercellularity. There are no deposits involving the peripheral glomerular capillary walls. (× 2000.) |
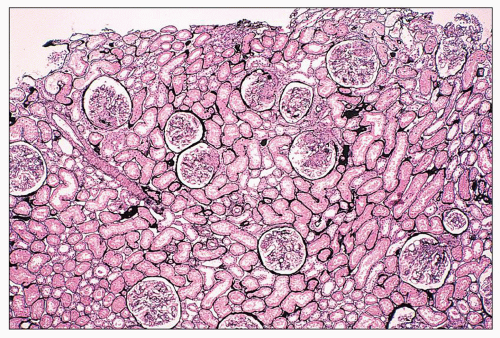 FIGURE 14.47 Lupus nephritis class III. A low-power view shows the focal and segmental distribution of the endocapillary proliferation, with some overlying crescents. Endocapillary proliferation affected less than 50% of the total glomeruli in this biopsy. (Jones methenamine silver stain; × 40.) |
pattern by light microscopy. The segments with endocapillary hypercellularity exhibit severe narrowing or occlusion of the glomerular capillaries by proliferation of endothelial and mesangial cells with a variable infiltration of mononuclear and polymorphonuclear leukocytes. This may be the only histologic finding in mild cases. More active examples may demonstrate any or all of the histologic features of activity previously discussed in a focal and segmental distribution. These include fibrinoid necrosis, karyorrhexis or pyknosis, rupture of the GBM, wire-loop deposits, intraluminal hyaline thrombi, and overlying cellular crescents (Fig. 14.49). Hematoxylin bodies are sometimes identified in the necrotizing lesions. Typically, foci of focal necrosis heal by progression to segmental or global scars with associated fibrous crescents or small synechiae to the Bowman capsule. Glomerular capillary walls in the “uninvolved” segments are usually thin and delicate.
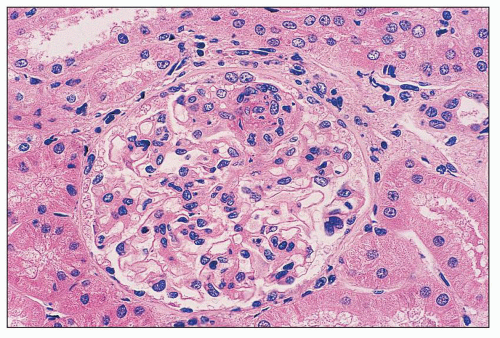 FIGURE 14.48 Lupus nephritis class III. There is segmental obliteration of glomerular capillary lumina by endocapillary proliferation, including infiltrating leukocytes, with associated fibrinoid necrosis. The adjacent lobules display mild mesangial hypercellularity. (H&E; × 400.) |
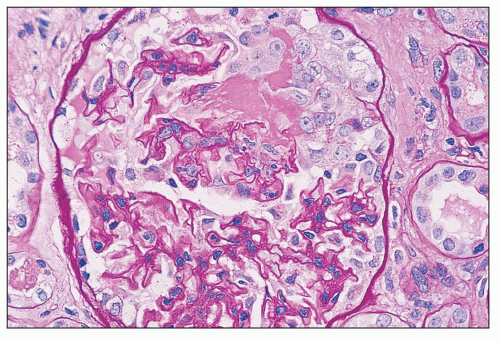 FIGURE 14.49 Lupus nephritis class III. There is segmental endocapillary proliferation and necrosis, with focal rupture of GBMs and an adjacent cellular crescent. (PAS, × 500.) |
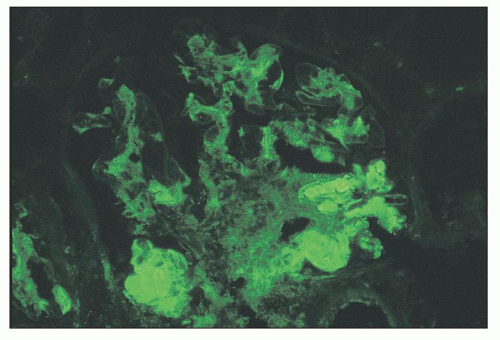 FIGURE 14.50 Lupus nephritis class III. The immunofluorescence micrograph shows segmental heavy IgG deposits in several glomerular capillary walls and lumina. The adjacent lobules have mesangial deposits. (× 500.) |
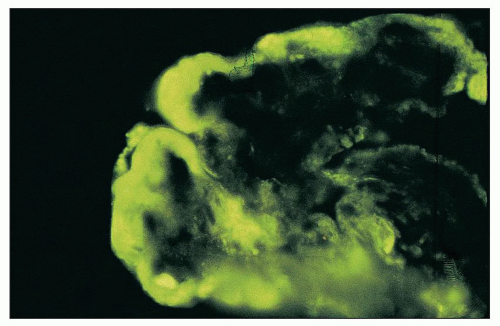 FIGURE 14.51 Wire-loop deposit. By immunofluorescence, there is a large subendothelial deposit that conforms to the contour of the GBM, producing a smooth comma-shaped outer contour. (× 1000.) |
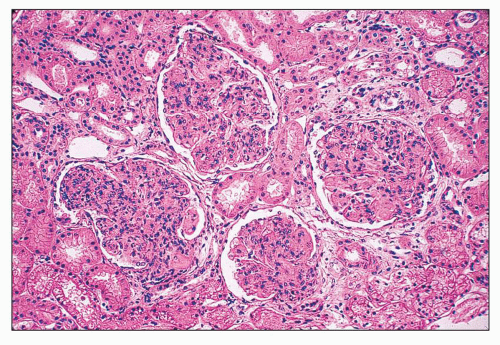 FIGURE 14.52 Lupus nephritis class IV-G. There is diffuse and global endocapillary proliferation involving all the glomeruli in this biopsy. (H&E, × 180). |
hypercellularity rather than predominantly segmental necrosis. All the lesions of active glomerular disease described for class III (e.g., nature of the endocapillary cells, fibrinoid necrosis, karyorrhexis and pyknosis, neutrophil infiltration, wire-loop deposits, hyaline thrombi, hematoxylin bodies, crescents) also apply to class IV. The two classes are distinguished by definition based on the percentage of glomeruli affected.
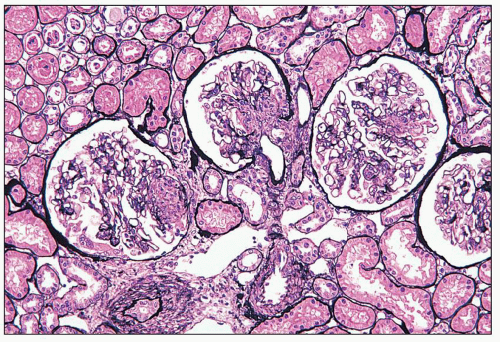 FIGURE 14.53 Lupus nephritis class IV-S. There is diffuse segmental glomerular proliferation involving more than 50% of the total glomeruli in this biopsy. (Jones methenamine silver, × 180.) |
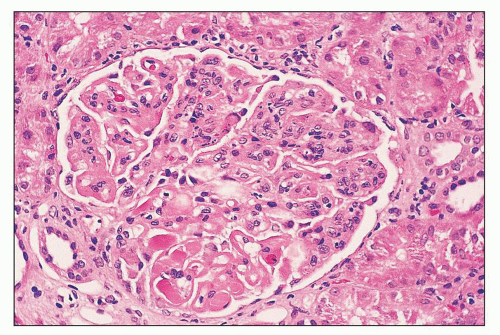 FIGURE 14.54 Lupus nephritis class IV. There is global narrowing of glomerular capillaries by mesangial and endocapillary proliferation. Wireloop deposits and hyaline thrombi are segmentally distributed. (H&E; × 500.) |
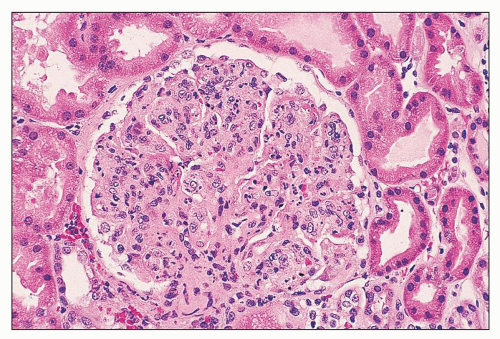 FIGURE 14.55 Lupus nephritis class IV. The endocapillary proliferation is global and includes many infiltrating neutrophils. (H&E, × 500.) |
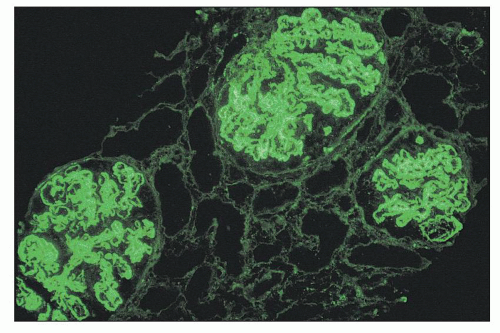 FIGURE 14.56 Lupus nephritis class IV. The low-power immunofluorescence micrograph shows intense, diffuse staining for IgG in the glomerular mesangium and peripheral capillary loops, consistent with a subendothelial distribution. (× 120.) |
pose difficulty in diagnosis (19,20). These include severe mesangial proliferative glomerulonephritis with diffuse subendothelial deposits and mesangiocapillary (or membranoproliferative) pattern glomerulonephritis and extensive subendothelial deposits with minimal glomerular hypercellularity (Fig. 14.58). Class IV-G disease has diffuse distribution of subendothelial deposits by immunofluorescence and electron microscopy, although the pattern of proliferation that accompanies these deposits varies considerably.
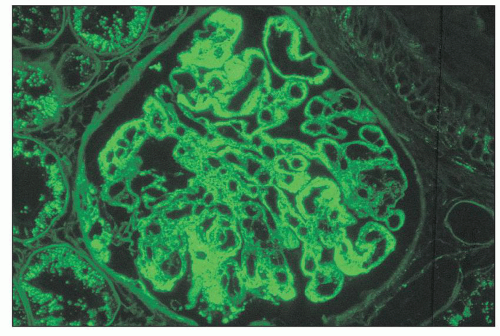 FIGURE 14.57 Lupus nephritis class IV. There are abundant deposits of IgG in the mesangium and the peripheral capillary walls. Most of the glomerular capillary wall deposits appear subendothelial, with more segmental subepithelial deposits. (Immunofluorescence micrograph, × 600.) |
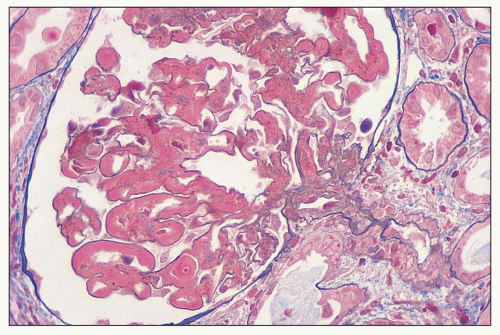 FIGURE 14.58 Lupus nephritis class IV. This example has diffuse wire-loop deposits without appreciable endocapillary proliferation. (Masson trichrome, × 600.) |
the tuft architecture (Fig. 14.61). However, more delicate semilinear staining for fibrinogen of about 1+ intensity may be seen more diffusely in areas of nonnecrotizing endocapillary proliferation in active glomerulonephritis.
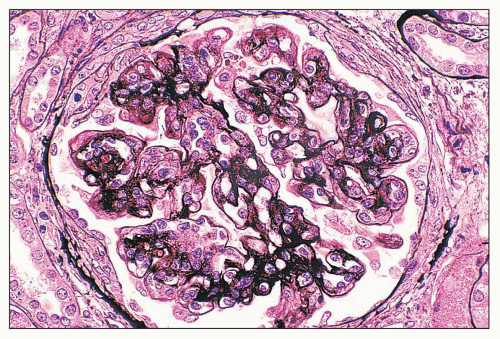 FIGURE 14.59 Lupus nephritis class IV. Membranoproliferative variant with lobular accentuation, nodular mesangial expansion, and numerous double contours of the GBM. (Jones methenamine silver; × 500.) |
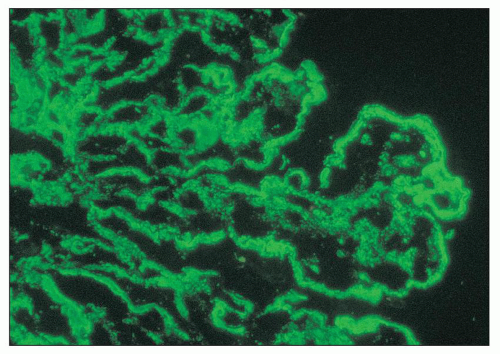 FIGURE 14.60 Lupus nephritis class IV. By immunofluorescence, granular deposits of C1q outline the mesangial areas and capillary loops. The peripheral capillary wall deposits are predominantly subendothelial, with minute, more irregular, subepithelial deposits. (× 700.) |
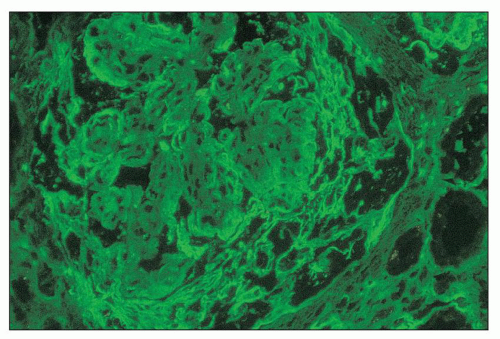 FIGURE 14.61 Lupus nephritis class IV. The immunofluorescence micrograph shows staining for fibrinogen in the periphery of the glomerular capillaries and the overlying crescent. (× 600.) |
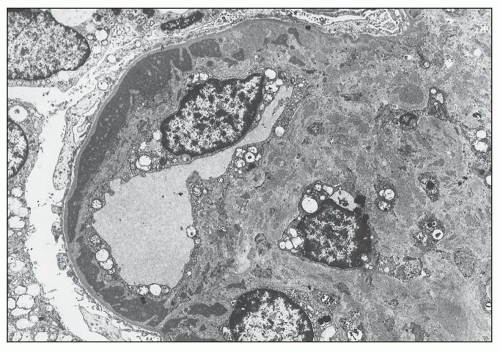 FIGURE 14.62 Lupus nephritis class IV. The electron micrograph shows a circumferential, subendothelial, electron-dense deposit that has been incorporated into the glomerular capillary wall by subendothelial neomembrane formation. There is marked mesangial expansion by mesangial proliferation and increased matrix containing many granular, electron-dense deposits. (× 2875.) |
steroid therapy, even when brief and inadequate to control the disease. Thus, the percentage of untreated patients with hypocomplementemia is considerably higher, 87% in one series (149). In untreated patients, the quantity of subendothelial deposits was found to correlate roughly with the reduction in serum C3 levels (209).
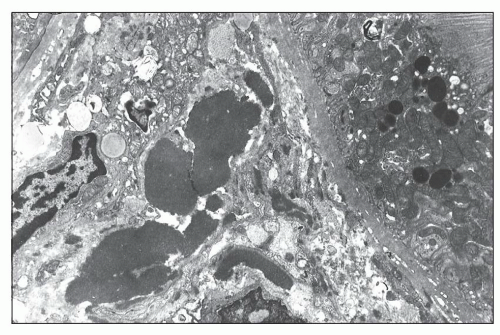 FIGURE 14.63 Lupus nephritis class IV. The electron micrograph from a fibrinoid necrotizing lesion shows endothelial necrosis with subendothelial deposition of abundant fibrillar fibrin and scant granular, electron-dense immune deposits. Neutrophils and monocytes infiltrate the glomerular capillary lumen. (× 2200.) |
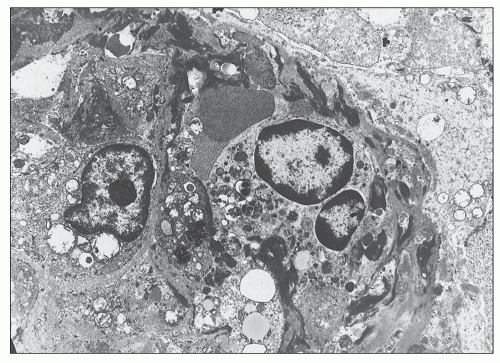 FIGURE 14.64 Lupus nephritis class IV. Massive electron-dense deposits are present in the interstitial collagen adjacentto a tubule. (× 2000.) |
a more global pattern of involvement over time (213). Hill et al. (214) studied 15 French patients with IV-S and 31 with IV-G and found that at baseline, patients with IV-G have more proteinuria, renal insufficiency, anemia, and hypocomplementemia. Morphologic differences include more membranoproliferative features, wire-loop deposits and hyaline thrombi, greater IF positivity involving the peripheral capillary walls, and less fibrinoid necrosis in IV-G than IV-S. Interestingly, repeat biopsies showed both types of interconversion (IV-S to IV-G in three patients and IV-G to IV-S in seven patients). The French group also observed no significant difference in 10-year survival between class IV-S and IV-G lesions at first biopsy (survival rates 65% vs. 60%, respectively). Interestingly, when repeat biopsies were performed 6 months following therapy, second biopsies with IV-G had a worse outcome than those with IV-S (214). A Japanese study also failed to find significant outcome differences in class IV-S versus IV-G (215). A recent meta-analysis (216) has analyzed the results of eight published studies addressing differences in IV-S versus IV-G (213,214,215,217,218,219,220,221) and concluded that there was no significant difference in renal outcome (doubling of serum creatinine or ESRD) between these histologic forms. These data call into question the validity of a classification that subcategorizes class IV based on the proportion of segmental and global lesions. Moreover, the IV-S category is relatively infrequent, comprising a minority of class IV biopsies (13% to 34%; see Table 14.8). Based on these data, future iterations of the ISN/RPS classification may opt to eliminate this distinction.
TABLE 14.8 Comparison of ISN/RPS class IV subgroups | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
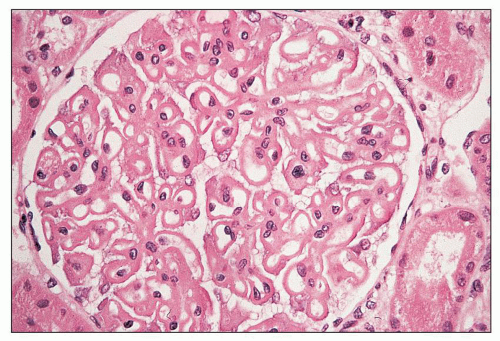 FIGURE 14.65 Lupus nephritis class V (modified Va). There is global thickening of glomerular capillary walls without mesangial proliferation. (H&E, × 500.) |
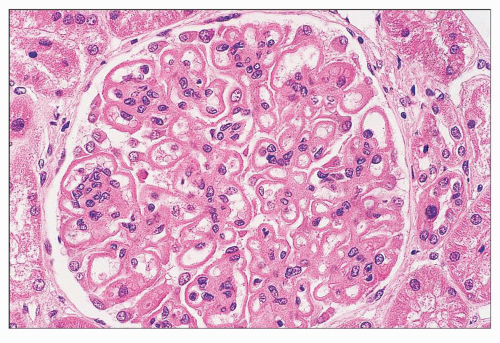 FIGURE 14.66 Lupus nephritis class V (modified Vb). There is regular thickening and rigidity of the glomerular capillary walls accompanied by global mesangial hypercellularity. (H&E; × 500.) |
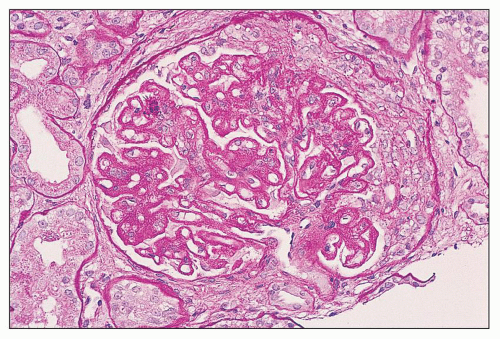 FIGURE 14.67 Lupus nephritis classes III and V (modified Vc). There is segmental endocapillary proliferation with an overlying crescent. The patent glomerular capillaries display chain-like thickenings of the glomerular capillary walls typical of membranous glomerulopathy. (PAS, × 400.) |
demonstrating that class Vd has an extremely poor prognosis, even worse than pure diffuse proliferative class IV (187,227,228). According to the ISN/RPS classification, as in the original WHO classification, the designation mixed class III and class V replaces the Vc lesion. Similarly, a designation of mixed class IV and class V replaces the Vd lesion. In the ISN/RPS schema, the additional designation of class V in the setting of class III or IV requires membranous involvement of at least 50% of glomerular capillary surface area of at least 50% of glomeruli by LM and/or IF.
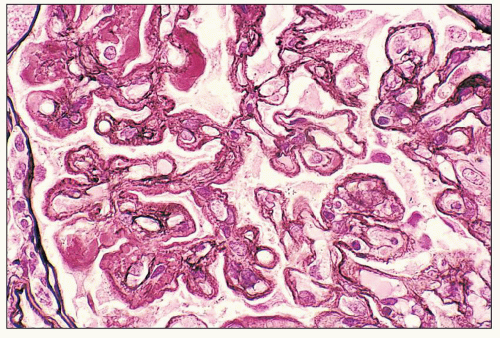 FIGURE 14.68 Lupus nephritis class IV and V (modified Vd). There is mixed, diffuse proliferative and membranous glomerulonephritis with complex thickening of the glomerular capillary walls by double contours enclosing subendothelial deposits and well-developed subepithelial spikes. (PAS-methenamine silver-Masson Ponceau stain, × 600.) |
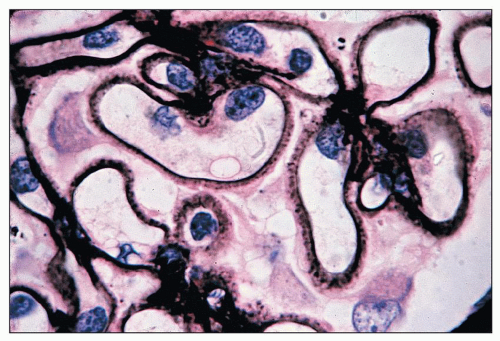 FIGURE 14.69 Lupus nephritis class V. Jones methenamine silver stain highlights the spiking of the GBMs. (× 1000.) |
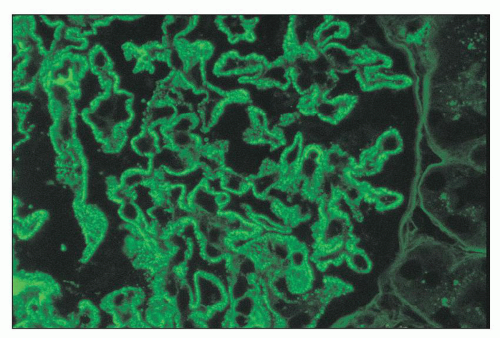 FIGURE 14.70 Lupus nephritis class V (modified Va). There are delicate subepithelial immune deposits staining for IgG. No mesangial deposits are observed. (Immunofluorescence micrograph, × 600.) |
also common but are not accompanied by endocapillary proliferation. Schwartz et al. (227) found them in eight of nine cases of lupus membranous nephritis (Va and Vb), and the Southwest Pediatric Nephrology Study Group found them in seven of nine cases (184). The presence of subendothelial deposits, which are rare in primary membranous glomerulopathy, is a particularly sensitive ultrastructural feature to distinguish membranous lupus nephritis from the primary form (158). Endothelial TRIs, a common feature of all ISN/RPS classes of lupus nephritis, are readily identified in class V. Tubulointerstitial deposits occur in approximately 25% of cases (64). Tubular atrophy and interstitial fibrosis usually parallel the distribution and severity of the glomerulosclerotic lesions.
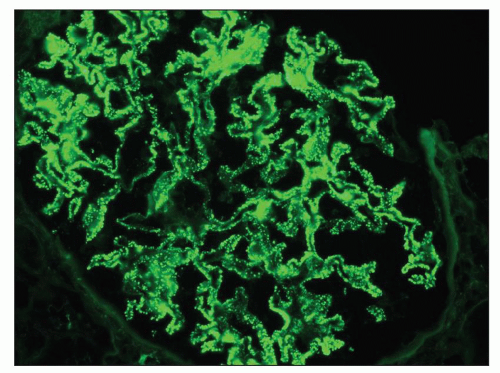 FIGURE 14.71 Lupus nephritis class V (modified Vb). There are heavy mesangial immune deposits of IgG with more delicate granular subepithelial deposits. (Immunofluorescence micrograph, × 600.) |
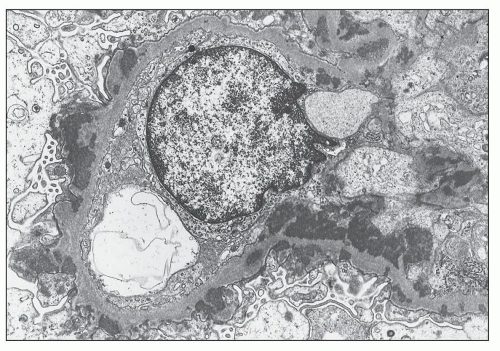 FIGURE 14.72 Lupus nephritis class Vb (membranous). The electron micrograph shows numerous subepithelial electron-dense deposits, some of which are surrounded by spiked projections of GBM. There are abundant mesangial electron-dense deposits. The glomerular capillary lumen is patent, and foot processes are extensively effaced. (× 3300.) |
subjecting the patient to radiopaque contrast materials, which are potentially nephrotoxic.
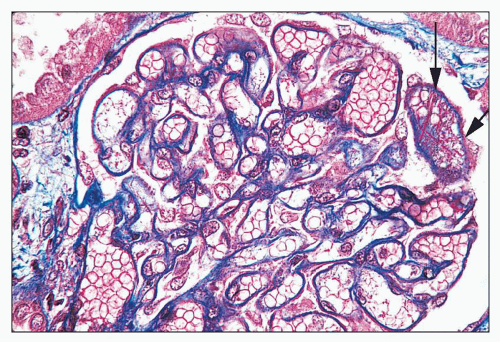 FIGURE 14.73 Lupus nephritis class V with superimposed RVT. Histologic features pointing to RVT include the separation of tubules by interstitial edema, marked glomerular capillary congestion, and fibrin strands within a glomerular capillary lumen (s). (Masson trichrome stain, × 500.) |
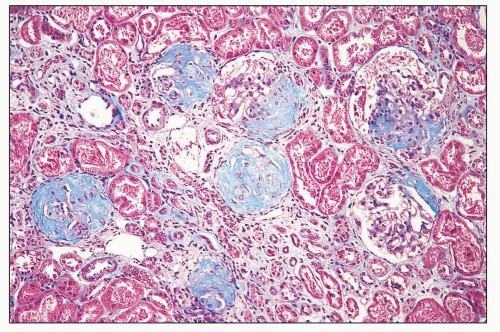 FIGURE 14.74 Lupus nephritis class VI. Extensive glomerular sclerosis shows vestiges of fibrous crescents. The global sclerosis affected more than 90% of glomeruli in this biopsy. Several glomeruli pictured here are segmentally sclerotic. Atrophic tubules alternate with groups of compensatorily hypertrophied tubules. (Masson trichrome, × 80.) |
TABLE 14.9 Class tranformations on repeat biopsies in 427 cases of lupus nephritis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
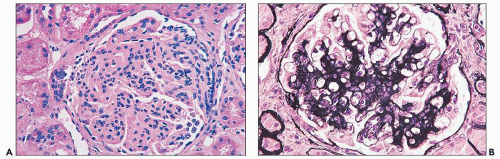 FIGURE 14.75 The initial renal biopsy of diffuse proliferative glomerulonephritis with marked endocapillary proliferation (A) regressed to a mesangial proliferative pattern with focal synechiae (B) following aggressive therapy. (A, H&E; B, Jones methenamine silver, × 500.) |
clinical improvement (249,250). It was presumed that there were some lesions, notably glomerular sclerosis, tubular atrophy, and interstitial fibrosis that are irreversible and may progress despite improvement in the proliferative and necrotic lesions. For more than five decades, investigators have attempted to analyze renal biopsy specimens of lupus nephritis with respect to active and chronic features as predictors of outcome and guides to therapy. The rationale is a simple one and based on the premise that active lesions are potentially treatable, whereas chronic lesions represent irreversible damage.
TABLE 14.10 Activity and chronicity indices | |||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||
research purposes, many consider it to be too intricate and time consuming to be applied to routine biopsy work-up.
areas of hypopigmentation. In SLE, lesions may occur on the face, scalp, neck and upper chest, or back. Pathologic examination of the skin reveals mononuclear inflammation of the dermis, follicular plugging, edema of the basal epidermis, and hyperkeratosis. Granular deposits of IgG and C3 are often detectable at the dermal-epidermal junction, corresponding to complexes of nucleosomes and antinucleosomal antibodies (266). Alopecia occurs in up to 70% of patients and may affect the eyebrows, eyelashes, beard, and scalp. Nail lesions, ridging and pitting in character, may also affect up to one fourth of SLE patients. Other skin lesions such as periungual erythema, telangiectasia, Raynaud phenomenon, and livedo reticularis are also common. The latter is particularly associated with a circulating lupus anticoagulant and/or APL antibodies. The occurrence of vasculitis may be manifested by splinter hemorrhages of the nailfold capillaries, small microinfarcts of the fingertips, and erythematous indurated lesions on the palmar thenar eminences (i.e., Janeway spots) and fingertips (i.e., Osler nodes).
in most patients with clinically significant kidney disease. In the 1980s, landmark studies from the NIH demonstrated that aggressive immunosuppression with cyclophosphamide in conjunction with corticosteroids was more effective than corticosteroids alone in producing remissions in most cases of lupus nephritis (280,281,282). However, this gain was accompanied by significant side effects from long-term immunosuppressive therapy, notably infections, gonadal toxicity, and bladder toxicity (283). Subsequent studies demonstrated that lower doses and shorter courses of cyclophosphamide, followed by maintenance therapy with mycophenolate mofetil (MMF) or azathioprine (AZA), were equally effective at preventing relapses and sustaining remissions as the higher doses of cyclophosphamide used in the original NIH protocols (284,285,286,287,288). More recently, MMF has emerged as a suitable alternative to cyclophosphamide for both proliferative and membranous lupus nephritis and may even be the treatment of choice in patients of African descent or Hispanic ethnicity (284,289,290,291,292,293). In addition, the use of calcineurin inhibitors in lupus nephritis is supported by several clinical studies (294,295,296). In the 21st century, novel biologic agents that target B-cell and T-cell function and cytokines have been employed in patients with SLE. Some of these drugs have entered the therapeutic armamentarium for lupus nephritis, whereas others remain under active study.
Related posts:
 Development of the Kidney
Development of the Kidney
 Membranous Glomerulonephritis
Membranous Glomerulonephritis
 IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
IgA Nephropathy and IgA Vasculitis (Henoch-Schönlein Purpura) Nephritis
 Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
Renal Involvement in Polyarteritis Nodosa, Kawasaki Disease, Takayasu Arteritis, and Giant Cell Arteritis
 Glomerular Diseases With Organized Deposits
Glomerular Diseases With Organized Deposits
 Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Pyelonephritis and Other Direct Renal Infections, Reflux Nephropathy, Hydronephrosis, Hypercalcemia, and Nephrolithiasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


