Drugs |
Normal Dosage |
% of Renal Excretion |
Dosage Adjustment in Renal Failure |
Comments |
HD |
CAPD |
CVVH |
GFR>50 |
GFR 10 to 50 |
GFR <10 |
Aminoglycoside Antibiotics |
|
|
|
|
|
Nephrotoxic, ototoxic
Toxicity worse when hyperbilirubinemic
Measure serum levels for efficacy and toxicity
Peritoneal absorption increases with presence of inflammation
Vd increases with edema, obesity, and ascites |
|
|
|
Streptomycin |
7.5 mg/kg q12h (1.0 g q24h for TB) |
60% |
q24h |
q24 to 72h |
q72 to 96h |
For the treatment of TB
May be less nephrotoxic than other members of class |
1/2 normal dose after dialysis |
20 to 40 mg/L/d |
Dose for GFR 10 to 50 and measure levels |
Kanamycin |
7.5 mg/kg q8h |
50% to 90% |
60% to 90% q12h or 100% q12 to 24h |
30% to 70% q12 to 18h or 100% q24 to 48h |
20% to 30% q24 to 48h or 100% q48 to 72h |
Do not use once-daily dosing in patients with creatinine clearance less than 30 to 40 mL/minutes or in patients with acute renal failure or uncertain level of kidney function |
1/2 full dose after dialysis |
15 to 20 mg/L/d |
Dose for GFR 10 to 50 and measure levels |
Gentamicin |
1.7 mg/kg q8h |
95% |
60% to 90% q8 to 12h or 100% q12 to 24h |
30% to 70% q12h or 100% q24 to 48h |
20% to 30% q24 to 48h or 100% q48 to 72h |
|
1/2 full dose after dialysis |
3 to 4 mg/L/d |
Dose for GFR 10 to 50 and measure levels |
Tobramicin |
1.7 mg/kg q8h |
95% |
60% to 90% q8 to 12h or 100% q12 to 24h |
30% to 70% q12h or 100% q24 to 48h |
20% to 30% q24 to 48h or 100% q48 to 72h |
|
1/2 full dose after dialysis |
3 to 4 mg/L/d |
Dose for GFR 10 to 50 and measure levels |
Netilmicin |
2 mg/kg q8h |
95% |
50% to 90% q8 to 12h or 100% q12 to 24h |
20% to 60% q12h or 100% q24 to 48h |
10% to 20% q24 to 48h or 100% q48 to 72 |
May be less ototoxic than other members of class
Peak 6 to 8, trough <2 |
1/2 full dose after dialysis |
3 to 4 mg/L/d |
Dose for GFR 10 to 50 and measure levels |
Amikacin |
7.5 mg/kg q12h |
95% |
60% to 90% q12h or 100% q12 to 24h |
30% to 70% q12 to 18h or 100% q24 to 48h |
20% to 30% q24 to 48h or 100% q48 to 72h |
Monitor levels
Peak 20 to 30, trough <5 |
1/2 full dose after dialysis |
15 to 20 mg/L/d |
Dose for GFR 10 to 50 and measure levels |
Cephalosporin |
|
|
|
|
|
Coagulation abnormalities, transitory elevation of BUN, rash, and serum sickness-like syndrome |
|
|
|
Oral Cephalosporin |
Cefaclor |
250 to 500 mg tid |
70% |
100% |
100% |
50% |
|
250 mg bid after dialysis |
250 mg q8 to 12h |
N/A |
Cefadroxil |
500 to 1 g bid |
80% |
100% |
100% |
50% |
|
0.5 to 1.0 g after dialysis |
0.5 g/d |
N/A |
Cefixime |
200 to 400 mg q12h |
85% |
100% |
100% |
50% |
|
300 mg after dialysis |
200 mg/d |
Not recommended |
Ceftibuten |
400 mg q24h |
70% |
100% |
100% |
50% |
|
300 mg after dialysis |
No data: Dose for GFR <10 |
Dose for GFR 10 to 50 |
Cefuroxime axetil |
250 to 500 mg tid |
90% |
100% |
100% |
100% |
Malabsorbed in presence of H2 blockers. Absorbed better with food. |
Dose after dialysis |
Dose for GFR <10 |
N/A |
Cephalexin |
250 to 500 mg tid |
95% |
100% |
100% |
100% |
Rare allergic interstitial nephritis. Absorbed well when given intraperitoneally. May cause bleeding from impaired prothrombin biosynthesis. |
Dose after dialysis |
Dose for GFR <10 |
N/A |
Cephradine |
250 to 500 mg tid |
100% |
100% |
100% |
50% |
|
Dose after dialysis |
Dose for GFR <10 |
N/A |
IV Cephalosporin |
Cefazolin |
1 to 2 g IV q8h |
80% |
q8h |
q12h |
q12 to 24h |
|
0.5 to 1.0 g after dialysis |
0.5 g q12h |
Dose for GFR 10 to 50 |
Cefepime |
1 to 2 g IV q8h |
85% |
q8 to 12h |
q12h |
q24h |
|
1 g after dialysis |
Dose for GFR <10 |
Not recommended |
Cefmetazole |
1 to 2 g IV q8h |
85% |
q8h |
q12h |
q24h |
|
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Cefoperazone |
1 to 2 g IV q12h |
20% |
No renal adjustment is required |
Displaced from protein by bilirubin. Reduce dose by 50% for jaundice. May prolong prothrombin time. |
1 g after dialysis |
None |
None |
Cefotaxime |
1 to 2 g IV q6 to 8h |
60% |
q8h |
q12h |
q12 to 24h |
|
1 g after dialysis |
1 g/d |
1g q12h |
Cefotetan |
1 to 2 g IV q12h |
75% |
q12h |
q12 to 24h |
q24h |
|
1 g after dialysis |
1 g/d |
750 mg q12h |
Cefoxitin |
1 to 2 g IV q6h |
80% |
q6h |
q8 to 12h |
q12h |
May produce false increase in serum creatinine by interference with assay |
1 g after dialysis |
1 g/d |
Dose for GFR 10 to 50 |
Ceftazidime |
1 to 2 g IV q8h |
70% |
q8h |
q12h |
q24h |
|
1 g after dialysis |
0.5 g/d |
Dose for GFR 10 to 50 |
Ceftriaxone |
1 to 2 g IV q24h |
50% |
No renal adjustment is required |
|
Dose after dialysis |
750 mg q12h |
Dose for GFR 10 to 50 |
Cefuroxime sodium |
0.75 to 1.5 g IV q8h |
90% |
q8h |
q8 to 12h |
q12 to 24h |
|
Dose after dialysis |
Dose for GFR <10 |
1.0 g q12h |
Penicillin |
|
|
|
|
|
Bleeding abnormalities, hypersensitivity. Seizures. |
|
|
|
Oral Penicillin |
Amoxicillin |
500 mg po tid |
60% |
100% |
100% |
50% to 75% |
|
Dose after dialysis |
250 mg q12h |
N/A |
Ampicillin |
500 mg po q6h |
60% |
100% |
100% |
50% to 75% |
|
Dose after dialysis |
250 mg q12h |
Dose for GFR 10 to 50 |
Dicloxacillin |
250 to 500 mg po q6h |
50% |
100% |
100% |
50% to 75% |
|
None |
None |
N/A |
Penicillin V |
250 to 500 mg po q6h |
70% |
100% |
100% |
50% to 75% |
|
Dose after dialysis |
Dose for GFR 10 |
N/A |
IV Penicillin |
Ampicillin |
1 to 2 g IV q6h |
60% |
q6h |
q8h |
q12h |
|
Dose after dialysis |
250 mg q12h |
Dose for GFR 10 to 50 |
Nafcillin |
1 to 2 g IV q4h |
35% |
No renal adjustment is required |
|
|
|
None |
None |
Dose for GFR 10 to 50 |
Penicillin G |
2 to 3 million units IV q4h |
70% |
q4 to 6h |
q6h |
q8h |
|
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Piperacillin |
3 to 4 g IV q4 to 6h |
No renal adjustment is required |
|
|
Sodium, 1.9 mEq/g |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Ticarcillin/clavulanate |
3.1 g IV q4 to 6h |
85% |
1 to 2 g q4h |
1 to 2 g q8h |
1 to 2 g q12h |
Sodium, 5.2 mEq/g |
3.0 g after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Piperacillin/tazobactam |
3.375 g IV q6 to 8h |
75% to 90% |
q4 to 6h |
q6 to 8h |
q8h |
Sodium, 1.9 mEq/g |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Quinolones |
|
|
|
|
|
Food, dairy products, tube feeding, and Al(OH)3 may decrease the absorption of quinolones. |
|
|
|
Ciprofloxacin |
200 to 400 mg IV q24h |
60% |
q12h |
q12 to 24h |
q24h |
Poorly absorbed with antacids, sucralfate, and phosphate binders. Intravenous dose 1/3 of oral dose. Decreases phenytoin levels. |
250 mg q12h (200 mg if IV) |
250 mg q8h (200 mg if IV) |
200 mg IV q12h |
Levofloxacin |
500 mg po qd |
70% |
q12h |
250 q12h |
250 q12h |
L-isomer of ofloxacin: appears to have similar pharmacokinetics and toxicities |
Dose for GFR <10 |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Moxifloxacin |
400 mg qd |
20% |
No renal adjustment is required |
|
No data |
No data |
No data |
Nalidixic acid |
1.0 g q6h |
High |
100% |
Avoid |
Avoid |
Agents in this group are malabsorbed in the presence of magnesium, calcium, aluminum, and iron. Theophylline metabolism is impaired. Higher oral doses may be needed to treat CAPD peritonitis. |
Avoid |
Avoid |
N/A |
Norfloxacin |
400 mg po q12h |
30% |
q12h |
q12 to 24h |
q24h |
See above. |
Dose for GFR <10 |
Dose for GFR <10 |
N/A |
Ofloxacin |
200 to 400 mg po q12h |
70% |
q12h |
q12 to 24h |
q24h |
See above. |
100 to 200 mg after dialysis |
Dose for GFR <10 |
300 mg/d |
Miscellaneous Agents |
Azithromycin |
250 to 500 mg po qd |
6% |
No renal adjustment is required |
No drug-drug interaction with CsA/FK |
None |
None |
None |
Clarithromycin |
500 mg po bid |
|
No renal adjustment is required |
|
None |
None |
None |
Clindamycin |
150 to 450 mg po tid |
10% |
No renal adjustment is required |
Increase CsA/FK level |
None |
None |
None |
Dirithromycin |
500 mg po qd |
|
No renal adjustment is required |
Nonenzymatically hydrolyzed to active compound erythomycylamine |
None |
No data: None |
Dose for GFR 10 to 50 |
Erythromycin |
250 to 500 mg po qid |
15% |
No renal adjustment is required |
Increase CsA/FK level, avoid in transplant patients |
None |
None |
None |
Ertapenem |
1 gm IV q24h |
|
1 gm IV q24h |
0.5 gm IV q24h |
0.5 gm IV q24h |
|
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR <30 |
Imipenem/Cilastatin |
250 to 500 mg IV q6h |
50% |
500 mg q8h |
250 to 500 q8 to 12h |
250 mg q12h |
Seizures in ESRD. Nonrenal clearance in acute renal failure is less than in chronic renal failure. Administered with cilastin to prevent nephrotoxicity of renal metabolite. |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Meropenem |
1 g IV q8h |
65% |
1 g q8h |
0.5 to 1g q12h |
0.5 to 1g q24h |
Fewer seizures compared to imipenem |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Metronidazole |
500 mg IV q6h |
20% |
No renal adjustment is required |
Peripheral neuropathy, increase LFTs, disulfiram reaction with alcoholic beverages |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Pentamidine |
4 mg/kg/day |
5% |
q24h |
q24h |
q48h |
Inhalation may cause bronchospasm, IV administration may cause hypotension, hypoglycemia, and nephrotoxicity |
None |
None |
None |
Trimethoprim/sulfamethoxazole |
800/160 mg po bid |
70% |
q12h |
q18h |
q24h |
Increase serum creatinine. Can cause hyperkalemia. |
Dose after dialysis |
q24h |
q18h |
Vancomycin |
1 g IV q12h |
90% |
q12h |
q24 to 36h |
q48 to 72h |
Nephrotoxic, ototoxic, may prolong the neuromuscular blockade effect of muscle relaxants
Peak 30 to 40. Trough 5 to 10. |
500 mg q12 to 24h (high FLX) |
1.0 gm q24 to 96h |
500 mg q12h |
Vancomycin |
125 to 250 mg po qid |
0% |
100% |
100% |
100% |
Oral vancomycin is indicated only for the treatment of C. diff. |
100% |
100% |
100% |
Antituberculosis Antibiotics |
Rifampin |
300 to 600 mg po qd |
20% |
No renal adjustment is required |
Decrease CsA/FK level. Many drug interactions. |
None |
Dose for GFR <10 |
Dose for GFR <10 |
Antifungal Agents |
Amphotericin B |
0.5 mg to 1.5 mg/kg/day |
<1% |
No renal adjustment is required |
Nephrotoxic, infusion related reactions, give 250 mL NS before each dose |
q24h |
q24h |
q24 to 36h |
Amphotec |
4 to 6 mg/kg/day |
< 1% |
No renal adjustment is required |
|
|
|
|
Abelcet |
5 mg/kg/day |
< 1% |
No renal adjustment is required |
|
|
|
|
AmBisome |
3 to 5 mg/kg/day |
< 1% |
No renal adjustment is required |
|
|
|
|
Azoles and other Antifungals |
|
|
|
|
|
Increase CsA/FK level |
|
|
|
Fluconazole |
200 to 800 mg IV qd/bid |
70% |
100% |
100% |
50% |
|
200 mg after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Flucytosine |
37.5 mg/kg |
90% |
q12h |
q16h |
q24h |
Hepatic dysfunction. Marrow suppression more common in azotemic patients. |
Dose after dialysis |
0.5 to 1.0 g/d |
Dose for GFR 10 to 50 |
Griseofulvin |
125 to 250 mg q6h |
1% |
100% |
100% |
100% |
|
None |
None |
None |
Itraconazole |
200 mg q12h |
35% |
100% |
100% |
50% |
Poor oral absorption |
100 mg q12 to 24h |
100 mg q12 to 24h |
100 mg q12 to 24h |
Ketoconazole |
200 to 400 mg po qd |
15% |
100% |
100% |
100% |
Hepatotoxic |
None |
None |
None |
Miconazole |
1,200 to 3,600 mg/day |
1% |
100% |
100% |
100% |
|
None |
None |
None |
Posaconazole |
200 mg qid |
1% |
100% |
100% |
100% |
|
|
|
|
Terbinafine |
250 mg po qd |
>1% |
100% |
100% |
100% |
Voriconazole |
4-6 mg/kg q12h |
1% |
100% |
100% |
100% |
Avoid IV formulation in CKD |
|
|
|
Caspofungin |
70 mg LD then 50 mg daily |
1% |
100% |
100% |
100% |
|
|
|
|
Micofungin |
100-150 mg IV daily |
1% |
100% |
100% |
100% |
|
|
|
|
Anidulafungin |
200 mg LD, then 100 mg daily |
1% |
100% |
100% |
100% |
|
|
|
|
Antiviral Agents |
Acyclovir |
200 to 800 mg po 5×/day |
50% |
100% |
100% |
50% |
Poor absorption. Neurotoxicity in ESRD. Intravenous preparation can cause renal failure if injected rapidly. |
Dose after dialysis |
Dose for GFR <10 |
3.5 mg/kg/d |
Adefovir |
10 mg q24h |
45% |
100% |
10 mg q48h |
10 mg q72 h |
Renal toxicity |
10 mg weekly after HD |
No data |
No data |
Amantadine |
100 to 200 mg q12h |
90% |
100% |
50% |
q96h to 7 days |
|
None |
None |
Dose for GFR 10 to 50 |
Cidofovir |
5 mg/kg weekly ×2 (induction); 5 mg/kg every 2 weeks |
90% |
Avoid in CKD |
No data: Avoid |
No data: Avoid |
Dose-limiting nephrotoxicity with proteinuria, glycosuria, renal insufficiency; nephrotoxicity and renal clearance reduced with coadministration of probenecid |
No data |
No data |
Avoid |
Delavirdine |
400 mg q8h |
5% |
No data: 100% |
No data: 100% |
No data: 100% |
|
No data: None |
No data |
No data: Dose for GFR 10 to 50 |
Didanosine |
200 mg q12h (125 mg if <60 kg) |
40% to 69% |
q12h |
q24h |
50% q24h |
Pancreatitis |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR <10 |
Emtricitabine |
200 mg q24h |
86% |
q24h |
q48-72h |
q 96 h |
|
|
Dose after dialysis |
No data |
Entecavir |
0.5 mg q24h |
62% |
q24h |
q48-72h |
q96 h |
|
|
Dose after dialysis |
No data |
Famciclovir |
250 to 500 mg po bid to tid |
60% |
q8h |
q12h |
q24h |
VZV: 500 mg po tid HSV: 250 po bid. Metabolized to active compound penciclovir. |
Dose after dialysis |
No data |
No data: Dose for GFR 10 to 50 |
Foscarnet |
40 to 80 mg IV q8h |
85% |
20 to 40 mg |
q8 to 24 h according to ClCr |
Nephrotoxic, neurotoxic, hypocalcemia, hypophosphatemia, hypomagnesemia, and hypokalemia |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Ganciclovir IV |
5 mg/kg q12h |
95% |
q12h |
q24h |
2.5 mg/kg qd |
Granulocytopenia and thrombocytopenia |
Dose after dialysis |
Dose for GFR <10 |
2.5 mg/kg q24h |
Ganciclovir |
1,000 mg po tid |
95% |
1,000 mg tid |
1,000 mg bid |
1,000 mg qd |
Oral ganciclovir should be used ONLY for prevention of CMV infection. Always use IV ganciclovir for the treatment of CMV infection. |
No data: Dose after dialysis |
No data: Dose for GFR <10 |
N/A |
Indinavir |
800 mg q8h |
10% |
No data: 100% |
No data: 100% |
No data: 100% |
Nephrolithiasis; acute renal failure due to crystalluria, tubulointerstitial nephritis |
No data: None |
No data: Dose for GFR <10 |
No data |
Lamivudine |
150 mg po bid |
80% |
q12h |
q24h |
50 mg q24h |
For hepatitis B |
Dose after dialysis |
No data: Dose for GFR <10 |
Dose for GFR 10 to 50 |
Maraviroc |
300 mg bid |
20% |
300 mg bid |
No data |
No data |
Drug interaction with CYP III-A |
No data |
No data |
No data |
Nelfinavir |
750 mg q8h |
No data |
No data |
No data |
No data |
|
No data |
No data |
No data |
Nevirapine |
200 mg q24h × 14d |
< 3 |
No data: 100% |
No data: 100% |
No data: 100% |
May be partially cleared by hemodialysis and peritoneal dialysis |
Dose after dialysis |
No data: Dose for GFR <10 |
No data: Dose for GFR 10 to 50 |
Oseltamivir |
75 mg bid |
99% |
75 bid |
75 mg daily |
75 mg q48h |
|
Dose after dialysis |
Ribavirin |
500 to 600 mg q12h |
30% |
100% |
100% |
50% |
Hemolytic uremic syndrome |
Dose after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Rifabutin |
300 mg q24h |
5% to 10% |
100% |
100% |
100% |
|
None |
None |
No data: Dose for GFR 10 to 50 |
Rimantadine |
100 mg po bid |
25% |
100% |
100% |
50% |
|
|
|
Ritonavir |
600 mg q12h |
3.50% |
No data: 100% |
No data: 100% |
No data: 100% |
Many drug interactions |
No data: None |
No data: Dose for GFR <10 |
No data: Dose for GFR 10 to 50 |
Saquinavir |
600 mg q8h |
<4% |
No data: 100% |
No data: 100% |
No data: 100% |
|
No data: None |
No data: Dose for GFR <10 |
No data: Dose for GFR 10 to 50 |
Stavudine |
30 to 40 mg q12h |
35% to 40% |
100% |
50% q12 to 24h |
50% q24h |
|
Dose for GFR <10 after dialysis |
No data |
No data: Dose for GFR 10 to 50 |
Telbivudine |
600 mg po daily |
|
100% |
600 mg q48h |
600 mg q96h |
|
Dose for GFR <10 after dialysis |
No data |
No data: Dose for GFR 10 to 50 |
Tenofovir |
300 mg q24h |
|
100% |
300 mg q48-72h |
300 mg q96h |
Nephrotoxic |
Dose for GFR <10 after dialysis |
No data |
No data: Dose for GFR 10 to 50 |
Valacyclovir |
500 to 1,000 mg q8h |
50% |
100% |
50% |
25% |
Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome |
Dose after dialysis |
Dose for GFR <10 |
No data: Dose for GFR 10 to 50 |
Valganciclovir |
900 mg po daily or bid |
|
100% |
50% |
25% |
Granulocytopenia and thrombocytopenia |
Dose after dialysis |
Dose for GFR <10 |
450 mg daily |
Vidarabine |
15 mg/kg infusion q24h |
50% |
100% |
100% |
75% |
|
Infuse after dialysis |
Dose for GFR <10 |
Dose for GFR 10 to 50 |
Zanamivir |
2 puffs bid × 5 days |
1% |
100% |
100% |
100% |
Bioavailability from inhalation and systemic exposure to drug is low |
None |
None |
No data |
Zalcitabine |
0.75 mg q8h |
75% |
100% |
q12h |
q24h |
|
No data: Dose after dialysis |
No data |
No data: Dose for GFR 10 to 50 |
Zidovudine |
200 mg q8h, 300 mg q12h |
8% to 25% |
100% |
100% |
100 mg q8h |
Enormous interpatient variation. Metabolite renally excreted. |
Dose for GFR <10 |
Dose for GFR <10 |
100 mg q8h |
HD, hemodialysis; CAPD, chronic peritoneal dialysis; CVVH, continuous venovenous hemofiltration; GFR, glomerular filtration rate; q, every; TB, tuberculosis; BUN, blood urea nitrogen; tid, three times a day; bid, twice a day; IV, intravenous; po, by mouth; ESRD, end-stage renal disease; C. diff, Clostridium difficile; CKD, chronic kidney disease; VZV, varicella zoster virus; HSV, herpes simplex virus; ClCr, creatinine clearance; CMV, cytomegalovirus. |
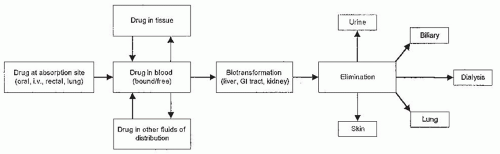


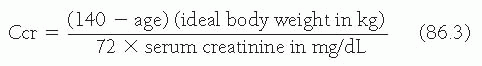
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Hypertension due to Primary Aldosteronism and Related Disorders
Hypertension due to Primary Aldosteronism and Related Disorders
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Immunobiology and Immunopharmacology of Renal Allograft Rejection
Immunobiology and Immunopharmacology of Renal Allograft Rejection



