Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Jan Novak
Bruce A. Julian
Immunoglobulin A nephropathy (IgAN), the most common primary glomerulonephritis in the world,1,2,3 is characterized by IgA-containing immune deposits in the glomerular mesangium. Berger and Hinglais described the disease in 1968 as a new entity based on the observation of “intercapillary deposits of IgA-IgG” using immunofluorescence examination of renal biopsy specimens from patients presenting with recurrent hematuria.4 Subsequently, it was established that the IgA deposits are exclusively of the IgA1 subclass.5 The same immunohistologic features are found in renal biopsy specimens from patients with Henoch-Schönlein purpura (HSP) and nephritis (HSPN) who have systemic findings of an IgA-associated vasculitis affecting the skin, gut, and joints.6,7,8,9 On the other hand, biopsies of clinically normal skin of some patients with IgAN have deposits of IgA in the walls of dermal capillaries.10 This finding, coupled with the shared biochemical abnormalities of circulating IgA1, has led to the postulate that the two diseases, IgAN and HSPN, represent the opposite ends of a spectrum of a disease process.9,11
IMMUNOGLOBULIN A NEPHROPATHY
Diagnosis
The histopathologic diagnosis of IgAN is usually clear and straightforward, with little need to consider a differential diagnosis. The key is the immunohistochemical identification of IgA deposition in the glomerular mesangium.
Immunofluorescence
A definitive diagnosis of IgAN can be made only by examination of renal cortical tissue with immunofluorescence microscopy or immunoperoxidase techniques. IgA is the dominant or codominant immunoreactant and is present predominantly in the mesangium, even in apparently normal or minimally affected glomeruli. Complement component C3 is usually found in the same distribution and is commonly accompanied by IgG, IgM, or both, although often with less intense fluorescence. Confocal microscopy has shown that when immune deposits show an outer layer of C3 rather than IgA, renal biopsy specimens exhibit more severe damage.12 C1q and C4 are found rarely; if present in substantial quantities, the possibility of lupus nephritis should be entertained.13
Capillary loop fluorescence for IgA is observed most frequently in patients with clinically active disease. Such biopsy specimens may also show fibrinogen in the mesangium and capillary walls and IgM in areas of glomerular sclerosis. Walls of small and medium-sized blood vessels may contain abundant granular C3.
Light Microscopy
The light microscopic hallmark of IgAN is expansion of mesangial area with proliferation of mesangial cells and increased extracellular matrix. In patients with mild disease, these changes may be quite focal and segmental. Some glomerular tufts may appear to be normal. Capillary loops usually are patent, with normal configuration of capillary walls. However, in more florid disease, mesangial proliferative activity results in the matrix extending peripherally and circumferentially in the capillary walls, resulting in a double-contouring or “tram-tracking” effect, usually with lumen narrowing. In active disease, there may be tuft necrosis associated with an exudate of fibrin and infiltration of neutrophils, some of which may show karyorrhexis. This feature is often associated with crescents in Bowman’s space.
In long-standing disease, areas of segmental tuft collapse and sclerosis, sometimes with overlying hyalinosis, are seen that usually are associated with broad synechiae. In progressive disease, the end result is glomerular obsolescence and sclerosis. All of these lesions may be found in one biopsy specimen. Focal segmental glomerulosclerosis may arise by several mechanisms, including postinflammatory scarring, compensatory hemodynamic changes after loss of nephrons, and primary damage to podocytes.14,15,16
Proportional to the degree of glomerular damage, there may be tubulointerstitial disease. When active glomerular disease is present, there often is interstitial edema associated with mild to moderate infiltrate of mononuclear cells and scattered neutrophils. Secondary tubular damage also may be evident. Interstitial scarring and tubular atrophy
are features of advanced disease.17 Hypertension-mediated damage may be seen patients with advanced disease. In an effort to standardize the description of the histologic features of the disease, an international consensus working group of nephrologists and nephropathologists has recently proposed a histologic classification scheme, the Oxford classification, based on light microscopic features.18,19
are features of advanced disease.17 Hypertension-mediated damage may be seen patients with advanced disease. In an effort to standardize the description of the histologic features of the disease, an international consensus working group of nephrologists and nephropathologists has recently proposed a histologic classification scheme, the Oxford classification, based on light microscopic features.18,19
Electron Microscopy
Ultrastructural studies show varying degrees of expansion and proliferation of mesangial cells and extracellular matrix, and electron-dense deposits of differing sizes and amounts in the matrix. Deposits are particularly common in paramesangial areas. Corresponding to the segmental nature of the disease process, the distribution and amount of deposits may be quite patchy. Some mesangial sites may be distinctly free of deposits, yet others in the same glomerulus may be packed with them. These deposits are usually solid and homogeneous. Electron-dense deposits are occasionally found in the subepithelial and subendothelial areas of the glomerular basement membranes. Deposits in the latter location may be associated with focal necrotizing glomerular lesions on light microscopy.
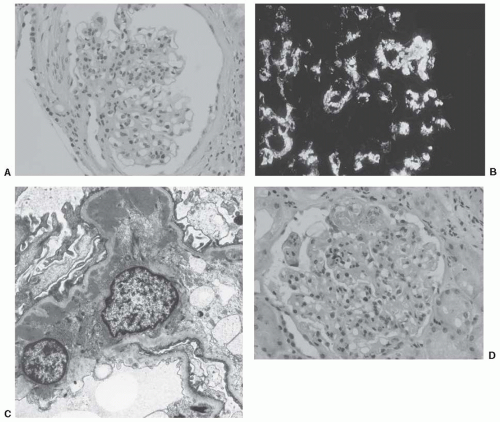 FIGURE 49.1 A: Glomerulus showing mesangial hypercellularity and increased mesangial matrix. (Hematoxylin and eosin stain, magnification, × 200.) B: Glomerulus, with brightly fluorescing mesangial deposits of IgA. (Fluorescinated antihuman IgA, magnification × 200.) C: Electron micrograph of glomerular mesangium showing multiple mesangial electron-dense deposits typical of IgA nephropathy. (Magnification × 3,000.) D: Glomerulus showing an acute lesion with segmental fibrinoid change and karyorhectic debris. (Hematoxylin and eosin, magnification × 200.) (continued) |
Nephrotic syndrome with the pathologic features of minimal change disease has been noted in a few patients with IgAN.20 In other patients, glomerular basement membranes are uniformly thin and this observation probably signifies the coexistence of two common conditions: IgAN and thin basement membrane nephropathy. However, one study of IgAN biopsies21 described 40% with thin glomerular basement membranes, so perhaps mesangial IgA deposits interfere in some way with synthesis of normal glomerular basement membranes. The histologic features of IgAN are illustrated in Figure 49.1,
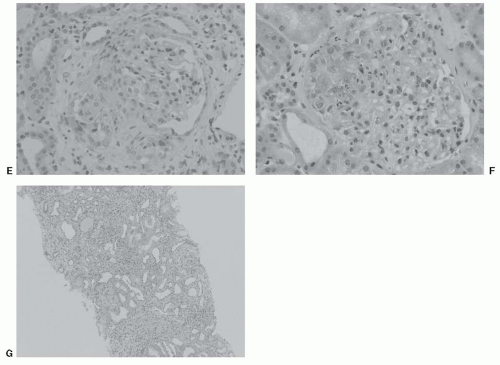 FIGURE 49.1 (Continued) E: Glomerulus showing an acute lesion with segmental crescent. (Hematoxylin and eosin, magnification × 200.) F: Sclerosing glomerulus with mesangial proliferation. (Hematoxylin and eosin, magnification × 200.) G: Low-power view of advanced IgAN, showing tubular dropout, interstitial fibrosis, and chronic inflammatory interstitial infiltrate. (Hematoxylin and eosin, magnification × 40.) (All photographs courtesy of Dr. James Nolan.) |
Pathogenesis
Kidney as Innocent Bystander
IgAN recurs frequently in renal allografts.22,23,24,25,26 Moreover, in isolated instances in which a kidney was transplanted from a donor with subclinical IgAN into a patient with non-IgAnephropathy renal disease, the immune deposits cleared from the affected kidney within several weeks.27 These clinical observations suggest that the cause of IgAN is extrarenal and there is considerable evidence indicating that the mesangial deposits originate from circulating IgA-containing immune complexes. It is well established that patients with IgAN frequently have elevated circulating levels of IgA and IgA-containing immune complexes.28,29,30 Idiotypic determinants are shared between the circulating complexes and the mesangial deposits31; however, a disease-specific idiotype has not been identified.32 Circulating immune complexes in patients with IgAN contain IgA1,28,33,34 the only IgA subclass in the mesangial immunodeposits.
Analysis of the glycosylation of IgA1 in patients with IgAN has yielded novel insights into the mechanisms underlying immune complex formation and mesangial deposition.30,34,35,36,37,38,39,40 Specifically, galactose deficiency in IgA1 O-glycans appears to be a key pathogenetic factor.36 Circulating complexes in patients with IgAN contain IgA1 with galactose-deficient hinge-region O-linked glycans.30,34,37,41 Notably, galactose-deficient IgA1 is the predominant glycosylation variant of IgA1 in the mesangial deposits, as determined by analyses of IgA1 eluted from glomeruli of nephrectomized kidneys or biopsy specimens from patients with IgAN.42,43 A relationship between galactose deficiency and nephritis also has been underscored by two other observations: (1) galactose-deficient IgA144 and IgA-IgG circulating complexes45 are found in sera of patients with HSPN but not in sera of patients with HSP and (2) patients with IgA1 myeloma have high circulating levels of IgA1, but only those with aberrantly glycosylated IgA1 develop immune-complex glomerulonephritis.46,47
Immunobiochemistry of IgA1: Galactose-deficient IgA1
IgA1 and IgA2 represent two structurally and functionally distinct subclasses of IgA in humans.48 IgA1 contains a unique hinge-region segment between the first and second constant-region domains of the heavy chains (Fig. 49.2A), with a high content of proline, serine, and threonine, that is the site of attachment of O-glycans. Up to six of the nine possible sites are occupied in each hinge region. These IgA1 O-glycans consist of N-acetylgalactosamine with a β1,3-linked galactose that may be sialylated.49,50,51,52,53,54 Sialic acid can be attached to N-acetylgalactosamine also
by an α2,6 linkage. The carbohydrate composition of the O-linked glycans in the hinge region of normal human serum IgA1 is variable (Fig. 49.2B). The prevailing glycans include galactose-N-acetylgalactosamine disaccharide and its mono- and di-sialylated forms.30,51,54,55,56,57,58 Galactose-deficient variants with terminal N-acetylgalactosamine or sialylated N-acetylgalactosamine are rarely found in the O-glycans of normal serum IgA1.51
by an α2,6 linkage. The carbohydrate composition of the O-linked glycans in the hinge region of normal human serum IgA1 is variable (Fig. 49.2B). The prevailing glycans include galactose-N-acetylgalactosamine disaccharide and its mono- and di-sialylated forms.30,51,54,55,56,57,58 Galactose-deficient variants with terminal N-acetylgalactosamine or sialylated N-acetylgalactosamine are rarely found in the O-glycans of normal serum IgA1.51
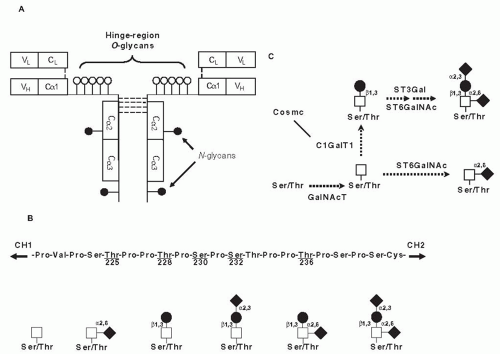 FIGURE 49.2 Structure and glycosylation of human IgA1. A: Monomer of IgA1 has two N-glycans and three to six O-glycans per heavy chain. B: Hinge-region amino acid sequence with common sites of O-glycan attachment and O-glycan variants of circulatory IgA1. The first two structures on the left side are galactose-deficient O-glycans. C: O-glycosylation of IgA1 is initiated by attachment of N-acetylgalactosamine to serine or threonine by UDP-N-acetylgalactosamine:polypeptideN-acetylgalactosaminyltransferases (GalNAc-Ts). The sites to be O-glycosylated and their order are determined by the specific set of GalNAc-Ts expressed in a particular cell type. N-acetylgalactosamine is then modified by addition of β1-3-linked galactose in a reaction catalyzed by UDP-galactose: N-acetylgalactosamine-α-Ser/Thr β,3-galactosyltransferase 1 (C1GalT1). Formation of active C1GalT1 depends on a specific chaperone, Cosmc. The N-acetylgalactosamine-galactose disaccharide can be further modified by attaching sialic acid to the galactose and/or N-acetylgalactosamine residues. |
The first step in the O-glycosylation of the IgA1 hinge region, with selection of the sites to be modified and the control of the sequence in this process, is accomplished by one of the few enzymes of the UDP-N-acetylgalactosamine:polypeptide N-acetylgalactosaminyltransferase family. In the second step, N-acetylgalactosamine is then modified by addition of β1-3-linked galactose, as detailed in Figure 49.2C. Lastly, one or both sugars can be further modified by the addition of sialic acid.59
In patients with IgAN, the basis of the aberrant O-glycosylation of IgA1 has been determined by using immortalized IgA1-secreting cell lines generated from circulating mononuclear cells. The galactose deficiency develops due to changes in expression and activity of key enzymes involved in O-glycosylation in the IgA1-producing cells (Fig. 49.2C).60 These changes in expression corresponded to decreased enzymatic activity of galactosyltransferase and elevated activity of sialyltransferase. Moreover, studies by other investigators identified mucosal, but not systemic, immune responses as the integral part of the aberrant O-glycosylation of IgA1.61,62 However, mechanisms involved in these pathways and their regulation remain to be elucidated.
Aggregates of IgA1 and Immune Complexes with Antibody Specific for Galactose-deficient IgA1
Normal circulatory IgA has a relatively short half-life (˜5 days) due to its rapid catabolism by hepatocytes.63 Hepatocytes express the asialoglycoprotein receptor52,64 that binds glycoproteins through terminal galactose or N-acetylgalactosamine residues.52,64,65 Because the structural prerequisite for binding is a terminal galactose or N-acetylgalactosamine, the absence or enzymatic removal of the otherwise terminal sialic acid is essential for effective binding.
Galactose-deficient IgA1 is retained in the circulation for prolonged intervals.66 Galactose deficiency in itself should not hinder disposal of IgA1 molecules because the asialoglycoprotein receptor recognizes terminal N-acetylgalactosamine as well as galactose.65 However, if the N-acetylgalactosamine is linked to sialic acid or is occupied by an antibody, it cannot be recognized by the receptor.41,67 Because galactose-deficient IgA1 is present primarily within immune complexes, it is plausible to speculate that this IgA1 does not effectively reach the hepatic asialoglycoprotein receptor. The larger size of the complexes, compared to that of uncomplexed IgA1, precludes binding to this receptor because the relatively small endothelial fenestrae block entry into the space of Disse. It is thus quite possible that immune complexes containing aberrantly glycosylated IgA1 are not efficiently cleared from the circulation and eventually deposit in the mesangium after passing through larger fenestrae in the glomerular capillaries.69,70,71,72 In animals, highmolecular-mass immune complexes induce more severe glomerular lesions than do small complexes.68
Mesangial Deposition of IgA1 and Inducement of Mesangial Injury
Immune complexes containing aberrantly glycosylated IgA1 can activate human mesangial cells in vitro, resulting in a proliferative response and overproduction of extracellular matrix components and cytokines/chemokines.37,41,73,74,75 Multiple studies have pointed to activation through an IgAspecific receptor(s) on mesangial cells. However, none of the known IgA receptors (CD89, asialoglycoprotein receptor, and polymeric immunoglobulin receptor) are expressed on human mesangial cells.37,41,76 Among the candidate receptors that may mediate binding of IgA1 and IgA1 complexes are CD71 (transferrin receptor)73,77,78 and the Fcα/µ receptor.79 CD71 appears to be the major IgA1 receptor on proliferating human mesangial cells.73,78,80 Notably, expression of CD71 is enhanced in the mesangia of IgAN patients and it colocalizes with IgA1 deposits.81 Engagement of CD71 by IgA1 induces cellular proliferation and cytokine production (e.g., interleukin [IL]-6, tumor growth factor [TGF]-β).80 This induction of cellular proliferation and cytokine production by IgA1 is inhibited completely by anti-CD71 antibody.80 It is not clear, however, whether CD71 is the only receptor or if any other receptor(s) plays a role in the activation of mesangial cells.
Although the signaling pathways and detailed mechanisms of the activation of mesangial cells by IgA1-containing immune complexes remain to be elucidated, it is generally agreed that mesangial cells represent the primary target in IgAN. There are two hypotheses for mechanisms leading to activation of mesangial cells by IgA1 in IgAN (Fig. 49.3). Both theories propose multiple hits and involvement of aberrantly glycosylated IgA1 and antiglycan antibodies. The first assumes formation of immune complexes in the circulation and their subsequent mesangial deposition and activation of mesangial cells (Fig. 49.3, solid lines).34,37,60,82 The other theory proposes that some of the aberrantly glycosylated IgA1 molecules are in the mesangium as lanthanic deposits, and later bound by newly generated antiglycan antibodies to form immune complexes in situ (Fig. 49.3, broken lines).83
Mesangial cells activated by immune complexes containing galactose-deficient IgA1 proliferate and overproduce extracellular matrix proteins, cytokines, and chemokines.37,39,73,74,78,84,85,86,87 These processes, when unchecked for substantial periods of time, may lead to expansion of the glomerular mesangium and, ultimately, glomerular fibrosis with loss of glomerular filtration function. Furthermore, humoral factors (e.g., tumor necrosis factor [TNF]-α and TGF-β)38,86,88,89 are released from mesangial cells activated by IgA1-containing immune complexes and alter podocyte gene expression and may thus increase glomerular permeability (Fig. 49.3). This mesangio-podocyte communication may be a mechanism to explain the occurrence of proteinuria and segmental glomerular sclerosis and tubulointerstitial injury in IgAN.14,86 Moreover, pathogenicity of IgA1 immune complexes may be enhanced in the presence of systemic signs of oxidative stress, and it has been hypothesized that oxidative stress may affect expression and progression of the disease.90
Genetic Influences
Genetic factors are known to play an important role in susceptibility to IgAN, as indicated by worldwide reports of extended multiplex pedigrees.91,92,93,94,95,96,97,98 Familial IgAN has offered a convenient tool for genetic studies and provided insight into the mechanism of inheritance. In some families, segregation of IgAN has been consistent with an autosomal-dominant transmission with incomplete penetrance. The incomplete penetrance, consistent with a complex-disease model, may reflect a requirement for additional genetic or environmental factors for clinical manifestation of the disease.
Gene mapping studies of diseases with complex determination are difficult and, thus far, no single mutation has been conclusively demonstrated to cause IgAN. Several genome-wide studies of familial IgAN have linked various loci with disease, including chromosomes 6q22-23 (named IGAN1), 4q26-31, 3p24-23, and 2q36.95,99,100 In contrast to the linkage studies, association studies involve a collection of sporadic cases and a group of unrelated controls. These studies can be performed for specific preselected genes (candidate-gene association studies) or on a genome-wide
scale to provide an unbiased examination of the genome, with the ability to detect and correct for population stratification. Replication of findings in independent cohorts is necessary for validation of both approaches.101
scale to provide an unbiased examination of the genome, with the ability to detect and correct for population stratification. Replication of findings in independent cohorts is necessary for validation of both approaches.101
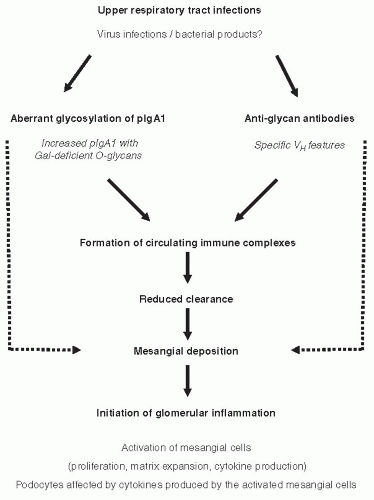 FIGURE 49.3 Mechanisms involved in IgAN pathogenesis. There are two hypotheses for mechanisms leading to the activation of mesangial cells by IgA1 in IgAN. Both theories propose multiple hits and involvement of aberrantly glycosylated IgA1 and antiglycan antibodies. The first assumes formation of immune complexes in the circulation and their subsequent mesangial deposition (solid lines).34,82 Galactose-deficient IgA1 produced by a population of IgA1-secereting cells60 is recognized by antiglycan antibodies with specific characteristics of variable region of the heavy chain82 and, consequently, immune complexes are formed from autoantigen (galactose-deficient IgA1) and autoantibody (glycanspecific antibody). It further assumes that some of the circulating complexes are pathogenic—that is, able to deposit in the mesangium and activate the resident mesangial cells.37 The other theory proposes that some of the aberrantly glycosylated IgA1 molecules are in the mesangium as lanthanic deposits, and later bound by newly generated antiglycan antibodies to form immune complexes in situ (broken lines).83 |
A single, unreplicated genome-wide association study (GWAS) in a small European cohort (533 cases) has reported association of IgAN with a human leukocyte antigen (HLA) locus.102 A recent replicated GWAS of a cohort of 3,144 IgAN cases of Chinese and European ancestry identified five loci.103 These loci explained up to a tenfold variation in interindividual risk and cumulatively accounted for 4% to 7% of the disease variance and included three independent loci in the major histocompatibility complex, a common deletion of CFHR1 and CFHR3 at chromosome 1q32 (complement factor H-related genes) and chromosome 22q12. The risk allele frequencies also strongly paralleled the prevalence of IgAN in the different populations. Furthermore, many of the IgAN-protective alleles imparted increased risk for other autoimmune or infectious diseases, suggesting complex selective pressures on allele frequencies.103 It is likely that studies with larger cohorts, and thus higher power, will define additional genetically influenced components in the pathogenesis of IgAN.
Recent studies of the glycosylation abnormalities of IgA1 offered prospects for a phenotypic biomarker for IgAN.104,105,106,107,108,109 A new quantitative lectin-binding assay enabled investigation of the inheritance of galactose-deficient IgA1 in familial and sporadic forms of IgAN.110 A high serum galactose-deficient IgA1 level was present in most index cases, as well as many of their first-degree relatives, whereas levels in spouses were indistinguishable from those in controls, eliminating an environmental effect. Segregation analysis of galactose-deficient IgA1 suggested inheritance of a major dominant gene with an additional polygenic component. The inheritance of galactose-deficient IgA1 has been confirmed in Chinese patients with familial and sporadic adult IgAN39,111 and in pediatric patients with IgAN and HSPN.94 Thus, aberrant IgA1 glycosylation is a common inherited defect that provides a unifying link in the pathogenesis of HSPN and familial and sporadic IgAN in many populations worldwide. GWAS studies using this new phenotype will likely provide information about the genetic and biochemical pathways leading to production of galactose-deficient IgA1 by IgA1-secreting cells.60 Furthermore, elevated circulatory levels of galactose-deficient IgA1 are antecedent to disease. However,
as most family members with elevated levels are asymptomatic, IgA1 glycosylation abnormalities are not sufficient to produce IgAN and additional cofactors, such as antiglycan antibodies,82 are required to trigger formation of pathogenic immune complexes (Fig. 49.3).
as most family members with elevated levels are asymptomatic, IgA1 glycosylation abnormalities are not sufficient to produce IgAN and additional cofactors, such as antiglycan antibodies,82 are required to trigger formation of pathogenic immune complexes (Fig. 49.3).
Clinical Disease
Incidence and Prevalence
The reported incidence and prevalence of IgAN varies widely, and depends, to some degree, on variations in criteria for renal biopsy in different countries. Studies from Europe have estimated the incidence at 15 to 40 new cases per million population per year. The incidence is higher in Japan and Korea,112,113 where screening for urinary abnormalities is routinely performed in school-aged children. Prevalence rates, expressed as a percentage of renal biopsy diagnoses, are reported to be 20% to 40% in Asia, Australia, Finland, and southern Europe. In the United States, the rate may be as low as 2% but in a large nephropathology referral practice IgAN accounted for 14% of nontransplant renal biopsies in adults aged 20 to 39 years.114




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
