rates are much higher, from threefold11,20 to 60-fold,12 in patients with even asymptomatic degrees of hypoosmolality compared to normonatremic patients. Although earlier studies associated increased mortality with serum [Na+] levels less than 130 mEq per L, more recent studies indicate an increased risk of mortality even when serum [Na+] levels decrease below 137 mEq per L.15 Remarkably, hyponatremia has been found to represent an independent predictor of worsened outcomes in virtually every disease ever studied, from congestive heart failure to tuberculosis to liver failure.16 Although this is probably because hypoosmolality is more an indicator of the severity of many underlying illnesses than it is an independent contributing factor to mortality, this presumption may not be true of all cases. These considerations emphasize the importance of a careful evaluation of all hyponatremic patients, regardless of the clinical setting in which they present.
measured serum [Na+] can simply be corrected by 1.6 to 2.4 mEq per L for each 100 mg per dL increase in serum glucose concentration greater than normal levels (100 mg per dL).
components of both solute depletion and water retention. Nonetheless, it is conceptually useful as a starting point for understanding the mechanisms underlying the pathogenesis of hypoosmolality and as a framework for discussions of therapy of hypoosmolar disorders.
TABLE 70.1 Pathogenesis of Hypoosmolar Disorders | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
of beer with little food intake for prolonged periods, called “beer potomania.”52,53 Although the volume of fluid ingested may not seem sufficiently excessive to overwhelm renal diluting mechanisms, in these cases free water excretion is limited by very low urinary solute excretion thereby causing water retention and dilutional hyponatremia. A reported case in which hyponatremia occurred in an ovolactovegetarian with a very low protein intake but no beer ingestion is consistent with this pathophysiologic mechanism.54 However, because most such patients have very low salt intakes as well, it is likely that relative depletion of body Na+ stores also is a contributing factor to the hypoosmolality in at least some cases.55
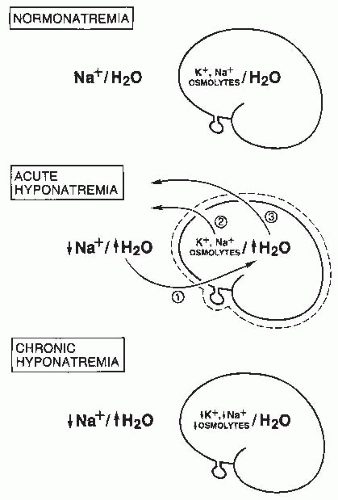 FIGURE 70.1 Schematic diagram of brain volume adaptation to hyponatremia. Under normal conditions brain osmolality and extracellular fluid (ECF) osmolality are in equilibrium (top panel; for simplicity the predominant intracellular solutes are depicted as K+ and organic osmolytes, and the extracellular solute as Na+). Following the induction of ECF hypoosmolality, water moves into the brain in response to osmotic gradients producing brain edema (dotted line, middle panel, #1). However, in response to the induced swelling the brain rapidly loses both extracellular and intracellular solutes (middle panel, #2). As water losses accompany the losses of brain solute, the expanded brain volume then decreases back toward normal (middle panel, #3). If hypoosmolality is sustained, brain volume eventually normalizes completely and the brain becomes fully adapted to the ECF hyponatremia (bottom panel). |
body undergo volume regulation during hypoosmolar conditions. However, volume regulatory processes are not limited to cells. Although most cases of hyponatremia clearly result from initial water retention induced by stimulated antidiuresis, it has always seemed likely that the resulting natriuresis served the purpose of regulating the volumes of the ECF and intravascular spaces. Many experimental and clinical observations are consistent with ECF volume regulation via secondary solute losses. First, dilutional decreases in concentrations of most blood constituents other than Na+ and Cl– do not occur in patients with SIADH,73 suggesting that their plasma volume is not nearly as expanded as would be predicted simply by the measured decreases in serum [Na+]. Second, an increased incidence of hypertension has never been observed in patients with SIADH,74 again arguing against significant expansion of the arterial blood volume. Third, results of animal studies in both dogs75 and rats76 have clearly indicated that a significant component of chronic hyponatremia is attributable to secondary Na+ losses rather than water retention. Furthermore, the relative contributions from water retention versus sodium loss vary with the duration and severity of the hyponatremia: water retention was found to be the major cause of decreased serum [Na+] in the first 24 hours of induced hyponatremia in rats, but Na+ depletion then became the predominant etiologic factor after longer periods (7-14 days) of sustained hyponatremia, particularly at very low (<115 mEq per L) serum [Na+] levels.76 Finally, multiple studies have attempted to measure body fluid compartment volumes in hyponatremic patients, but without consistent results that indicate either plasma or ECF volume expansion.1,57,77,78 In particular, a report of body fluid space measurements using isotope dilution techniques in hyponatremic and normonatremic patients with small cell lung carcinoma showed no differences between the two groups with regard to exchangeable sodium space, ECF volume by 35SO4 distribution, or total body water.79 Such results have traditionally been explained by the relative insensitivity of isotope dilution techniques for measurement of body fluid compartment spaces, but an equally plausible possibility is that in the chronically adapted hyponatremic state body fluid compartments have regulated their volumes back toward normal via a combination of extracellular (predominantly electrolyte) and intracellular (electrolyte and organic osmolyte) solute losses.80 Figure 70.2 schematically illustrates some of the volume regulatory processes that likely occur in response to water retention induced by inappropriate antidiuresis. The degree to which solute losses versus water retention contribute to the resulting hyponatremia will vary in association with many different factors, including the etiology of the hyponatremia, the rapidity of development of the hyponatremia, the chronicity of the hyponatremia, the volume of daily water loading and subsequent volume expansion, and undoubtedly some degree of individual variability as well. It therefore hardly seems surprising that studies of hyponatremic patients have failed to yield uniform results regarding the pathogenesis of hyponatremia in view of the marked diversity of hyponatremic patients and their presentation at different times during the process of adaptation to hypoosmolality via volume regulatory processes.
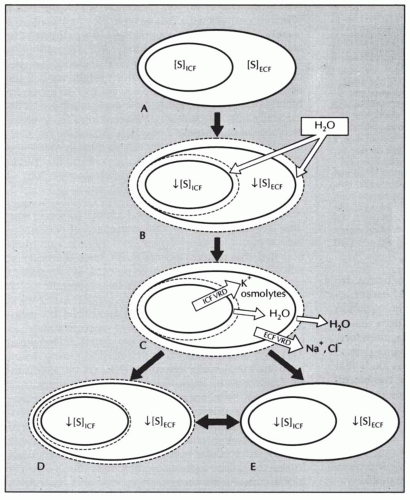 FIGURE 70.2 Schematic illustration of potential changes in whole body fluid compartment volumes at various times during adaptation to hyponatremia. Under basal conditions the concentration of effective solutes in the extracellular fluid ([S]ECF) and the intracellular fluid ([S]ICF) are in osmotic balance (A). During the first phase of water retention resulting from inappropriate antidiuresis the excess water distributes across total body water, causing expansion of both ECF and ICF volumes (dotted lines) with equivalent dilutional decreases in [S]ICF and [S]ECF (B). In response to the volume expansion, compensatory volume regulatory decreases (VRD) occur to reduce the effective solute content of both the ICF (via increased electrolyte and osmolyte extrusion mediated by stretch activated channels and down-regulation of synthesis of osmolytes and osmolyte uptake transporters) and the ECF (via pressure diuresis and natriuretic factors) (C). If both processes go to completion, such as under conditions of fluid restriction, a final steady state can be reached in which ICF and ECF volumes have returned to normal levels but [S]ICF and [S]ECF remain low (E). In most cases this final steady state is not reached, and moderate degrees of ECF and ICF expansion persist, but significantly less than would be predicted from the decrease in body osmolality (D). Consequently, the degree to which hyponatremia is due to dilution from water retention versus solute depletion from volume regulatory processes can vary markedly depending on which phase of adaptation the patient is in, and also on the relative rates at which the different compensatory processes occur (e.g., delayed ICF VRD can worsen hyponatremia due to shifts of intracellular water into the extracellular fluid as intracellular organic osmolytes are extruded and subsequently metabolized, likely accounting for some component of the hyponatremia unexplained by the combination of water retention and sodium excretion in previous clinical studies). (From Verbalis JG. Hyponatremia: epidemiology, pathophysiology, and therapy. Curr Opin Nephrol Hyperten. 1993;2:636-652, with permission.) |
indistinguishable in terms of water balance.90,91 Hyponatremia from diuretic use also can present without clinically evident hypovolemia, and the urine [Na+] will often be elevated in such cases because of the renal tubular effects of the diuretics.38 Recent studies have suggested that the fractional excretion of uric acid may provide a better measure of ECF volume status in hyponatremic patients on diuretics.92 A low urine [Na+] suggests a depletion-induced hypoosmolality from ECF losses with subsequent volume replacement by water or other hypotonic fluids. The solute loss often is generally nonrenal in origin, but an important exception is recent cessation of diuretic therapy, because urine [Na+] can quickly decrease to low values within 12 to 24 hours after discontinuation of the diuretic. The presence of a low serum [K+] is an important clue to diuretic use, because few of the other disorders that cause hypoosmolality are associated with significant hypokalemia. However, even in the absence of hypokalemia, any hypoosmolar, clinically euvolemic patient taking diuretics should be assumed to have solute depletion and treated accordingly; subsequent failure to correct the hypoosmolality with isotonic saline administration and persistence of an elevated urine [Na+] after discontinuation of diuretics then requires reconsideration of a diagnosis of dilutional hypoosmolality. A low urine [Na+] also can also be seen in some cases of hypothyroidism, in the early stages of decreased EABV before the development of clinically apparent sodium retention and fluid overload, or during the recovery phase from SIADH. Hence, a low urine [Na+] is less meaningful diagnostically than is a high value.
TABLE 70.2 Differential Diagnosis of Hyponatremia | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
findings that support this diagnosis. Several points about each of these criteria deserve emphasis and/or qualification:
TABLE 70.3 Criteria for the Diagnosis of SIADH | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
to assess water excretion after treatment of an underlying disorder thought to be causing SIADH. For example, after discontinuation of a drug associated with SIADH in a patient who has already achieved a normal plasma osmolality by fluid restriction, a normal water load test can confirm the absence of persistent inappropriate antidiuresis much more quickly than can simple monitoring of the serum [Na+] during a period of ad libitum fluid intake. Despite these limitations as a diagnostic clinical test, the water load test remains an extremely useful tool in clinical research for quantitating changes in free water excretion in response to physiologic or pharmacologic manipulations.
it would be preferable to establish a diagnosis before making a decision regarding treatment options. Nonetheless, in difficult cases of euvolemic hypoosmolality, an appropriate therapeutic response can sometimes be helpful in confirming a diagnosis of SIADH.
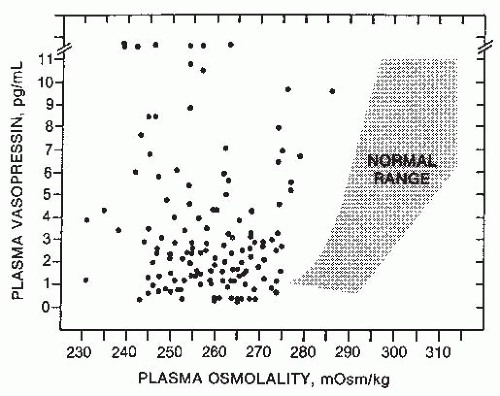 FIGURE 70.3 Plasma AVP levels in patients with SIADH as a function of plasma osmolality. Each point depicts one patient at a single point in time. The shaded area represents AVP levels in normal subjects over physiologic ranges of plasma osmolality. The lowest measurable plasma AVP levels using this radioimmunoassay was 0.5 pg per mL. (From Robertson GL, Aycinena P, Zerbe RL. Neurogenic disorders of osmoregulation. Am J Med. 1982;72:339-353, with permission.) |
regulatory and emetic centers (Fig. 70.4). Although various components of these pathways have yet to be fully elucidated, many of them appear to have inhibitory as well as excitatory components.109 Consequently, any diffuse CNS disorder can potentially cause AVP hypersecretion either by nonspecifically exciting these pathways via irritative foci, or alternatively by disrupting them and thereby decreasing the level of inhibition impinging upon the AVP neurons in the neurohypophysis. The wide variety of diverse CNS processes that can potentially cause SIADH stands in contrast to CNS causes of diabetes insipidus, which are for the most part limited to lesions localized to the hypothalamus and/or posterior pituitary that destroy the magnocellular vasopressin neurons (see Chapter 71).
TABLE 70.4 Common Etiologies of SIADH | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
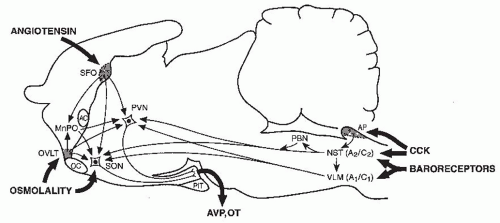 FIGURE 70.4 Diagrammatic summary of the primary brain pathways mediating AVP secretion in response to the major factors that stimulate pituitary AVP secretion. Osmolality activates neurons throughout the anterior hypothalamus, including the SFO and MnPO, but the OVLT appears to be uniquely sensitive to osmotic stimulation and is essential for osmotically stimulated AVP and OT secretion; in addition, osmotic stimulation can act directly on magnocellular neurons which themselves are intrinsically osmosensitive. Similarly, circulating angiotensin II activates cells throughout the OVLT and MnPO, but the SFO appears to be its major and essential site of action. For both of these stimuli, projections from the SFO and OVLT to the MnPO activate both excitatory and inhibitory interneurons that project to the SON and PVN and modulate the direct circumventricular inputs to these areas. Emetic stimuli act both on gastric vagal afferents which terminate in the NST and in some cases also act directly at the AP. Most of the AVP secretion appears to be a result of monosynaptic projections from catecholaminergic A2/C2 cells in the NST. Baroreceptor-mediated stimuli such as hypovolemia and hypotension are considerably more complex. Although they also arise from cranial nerves (IX and X) which terminate in the NST, most experimental data suggest that the major projection to magnocellular AVP neurons arises from catecholaminergic A1 cells of the VLM that are activated by excitatory interneurons from the NST, although some component might also arise from multisynaptic projections through other areas such as the PBN.AC, anterior commissure; AP, area postrema; AVP, arginine vasopressin; MnPO, median preoptic nucleus; NST, nucleus of the solitary tract; OC, optic chiasm; OT, oxytocin; OVLT, organum vasculosum of the lamina terminalis; PBN, parabrachial nucleus; PIT, anterior pituitary; PVN, paraventricular nucleus; SFO, subfornical organ; SON, supraoptic nucleus; VLM, ventrolateral medulla. |
effects on oxytocin rather than AVP secretion.115 Some studies of SSRIs in humans have failed to show significant effects on AVP secretion,116 although others support this mechanism.117 However, hyponatremia following SSRI administration has been reported almost exclusively in the elderly, at rates as high as 22% to 28% in some studies,118,119,120 although larger series have suggested an incidence closer to 1 in 200.121 This therefore suggests the possibility that elderly patients are uniquely hypersensitive to serotonin stimulation of AVP secretion. A similar effect is likely also responsible for the recent reports of severe fatal hyponatremia caused by use of the recreational drug 3,4-methylenedioxymethamphetamine, “ecstasy,”122,123 because this agent also possesses substantial serotonergic activity.124 Studies of cFos expression in rats indicate that ecstasy appears to activate hypothalamic magnocellular neurons,125 suggesting direct effects on AVP secretion as the etiology of the SIADH, and recent studies in humans support this mechanism.126,127
and primarily in psychiatric patients who have several other potential causes of inappropriate AVP secretion.39,171,172 This is in part because of chronic adaptation to the effects of nicotine, but also because the short half-life of AVP in plasma (approximately 15 min in humans173) limits the duration of antidiuresis produced by relatively short-lived stimuli such as smoking. Although nausea remains the most potent stimulus to AVP secretion known in man,174 chronic nausea is rarely associated with hypoosmolality unless accompanied by vomiting with subsequent ECF solute depletion followed by ingestion of hypotonic fluids.175 Similar to smoking, this is probably attributable to the short half-life of AVP, but also to the fact that most such patients are not inclined to drink fluids under such circumstances. However, hyponatremia can occur when such patients are infused with high volumes of hypotonic fluids. This is likely a factor contributing to the hyponatremia that often occurs in cancer patients who are receiving chemotherapy.103 Finally, a causal relation between stress and SIADH has often been suggested, but never conclusively established. This underscores the fact that stress, independent of associated nausea, dehydration, or hypotension, is not a major stimulus causing sustained elevations of AVP levels in humans.176
produce hypoosmolality when exogenous fluid intakes are high, as in psychiatric patients with polydipsia.189 Recent studies of patients with SIADH and hypopituitarism have measured high nonsuppressible levels of urinary aquaporin-2 excretion that correlated with their impaired water excretion, supporting persistent activation of AVP V2 receptors as the cause of the water retention.190
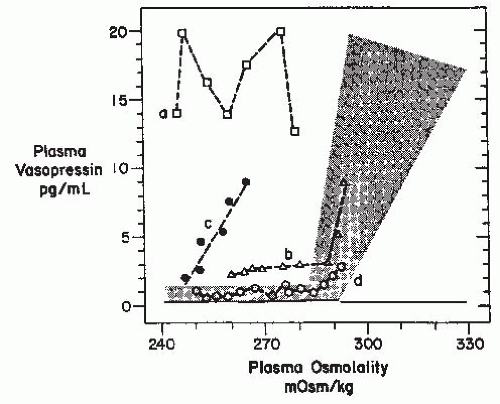 FIGURE 70.5 Schematic summary of different patterns of AVP secretion in patients with SIADH. Each line (a-d) represents the relation between plasma AVP and plasma osmolality of individual patients in whom osmolality was increased by infusion of hypertonic NaCl. The shaded area represents plasma AVP levels in normal subjects over physiologic ranges of plasma osmolality. (From Robertson GL. Thirst and vasopressin function in normal and disordered states of water balance. J Lab Clin Med. 1983;101:351-371, with permission.) |
or exaggerate such hypovolemic conditions, potentially accounting for the occurrence of a reset osmostat pattern of AVP secretion found in some patients with cancers. Recent reports of a 3% to 4% incidence of SIADH in patients with advanced head and neck malignancies represents a group in which some, although clearly not all,203 of the hyponatremia might also be secondary to interference with vagal baroreceptor pathways.204 However, not all cases of SIADH can be comfortably ascribed to nonosmotic stimuli because it is difficult to identify any such possible stimuli in many patients. Another possibility is that brain pathways conveying afferent signals that actively inhibit AVP secretion from hypothalamic magnocellular neurons may be impaired in some patients. Substantial data support the likelihood that hypoosmolality does not simply lead to decreased AVP secretion by virtue of absence of excitatory osmoreceptor inputs, but rather represents a state of active inhibition of the AVP-secreting neurons,205 possibly via endogenous opioid206 or gammaamino butyric acid (GABA) pathways.109,207 In this case, it would be easy to imagine that impairments or alterations in the activity of these inhibitory pathways might allow continued AVP secretion despite hypoosmolality. Although such abnormalities have not yet been identified, there is one clinical situation in which a decreased inhibitory tone to AVP neurons does clearly lead to enhanced AVP secretion: elderly patients have decreased AVP responses to orthostasis but exaggerated responses to osmotic stimuli.208,209 The latter is presumably due to a diminution of inhibitory, as well as excitatory, inputs from brainstem baroreceptive centers to the hypothalamus, thereby producing an unopposed stimulation by osmotic stimuli from the anterior hypothalamus (Fig. 70.4). This phenomenon could contribute to the unusually high frequency of SIADH seen in elderly individuals.7,13,14,163
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


