SYSTEMIC SCLEROSIS (SCLERODERMA)
Scleroderma is a broad, often confusing, term that encompasses a subset of chronic connective tissue diseases resulting from the overproduction and accumulation of collagen and other extracellular matrix proteins. Derived from the Greek word
sklēros, meaning hard, and
derma, meaning skin, scleroderma describes the hardened skin that is the hallmark clinical feature of this disorder. However, the disease is far more complex than this term implies. As such, scleroderma is now classified into two accepted disease subsets, morphea and systemic sclerosis (SSc), which more accurately reflect the broad range of clinical features seen in this disease.
1,
2 Morphea, the localized form of scleroderma, is limited to the skin and generally carries a favorable prognosis with normal life expectancy. On the other hand, SSc, a widespread disorder with internal organ involvement, is generally associated with a worse prognosis and significant disability and mortality.
SSc is characterized by intense uncontrolled fibrosis of the skin, subcutaneous tissues, and organs (most notably the kidneys, lungs, gastrointestinal tract, and heart), accompanied by a proliferative and obliterative vasculopathy. There is considerable heterogeneity in the clinical manifestations and severity of SSc. Two well-recognized clinical subsets of SSc, the limited cutaneous (lcSSc) and diffuse cutaneous (dcSSc) forms, are used to further subclassify patients based on the extent and distribution of skin involvement. Autoantibody profiles, patterns of internal organ involvement, and prognosis differ considerably between the groups. The limited form, which primarily affects the skin distal to the elbows and knees, is typically associated with Raynaud phenomenon, telangiectasia, and gastrointestinal involvement as part of the CREST syndrome (calcinosis cutis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia). Pulmonary hypertension and anticentromere antibodies are more common in this disease subset. Diffuse SSc is associated with more extensive sclerosis involving the skin proximal to the elbow and knee flexures or the trunk and earlier onset of internal organ involvement. Antitopoisomerase I and anti-RNA polymerase III antibodies are frequently present. Patients with dcSSc are at increased risk of developing the devastating complication, scleroderma renal crisis, discussed in the following paragraphs.
Epidemiology
Although there are conflicting estimates of prevalence and incidence of SSc, it is known that it is relatively uncommon compared to other connective tissue diseases. Prevalence estimates range from 7 to 489 cases per million persons and an incidence range from 0.6 to 122 cases per million persons per year (for comparison, prevalence of lupus ranges from 200 to 1500 cases per million persons).
3,
4 The wide range in reported estimates between studies reflects a multitude of factors including different diagnostic definitions, inclusion of different subtypes of disease, data acquisition strategies, geographic variation, and study methods and design. The average disease onset is between the fourth and fifth decades. There is a female predominance with a female to male ratio ranging from 3:1 to 8:1, and it is almost twice as common in blacks as in whites.
Pathogenesis
The events that lead to the systemic fibrosis, microvascular damage, and immune dysregulation that are characteristic of SSc are only partially understood. Collectively, the body of evidence supports a multistep process involving a complex interplay between the vascular system, the immune system, and the extracellular matrix. This occurs in the context of a unique genetic susceptibility
5 and possible environmental stimuli.
6,
7 Although a detailed discussion of disease pathogenesis is beyond the scope of this chapter, important concepts will be highlighted here. For a more detailed discussion, we refer the reader to several recent excellent reviews.
8,
9,
10,
11Injury to the vascular endothelium, primarily of small vessels, is an important proximal event preceding the development of fibrosis.
12 The inciting factor remains unknown, though numerous environmental and infectious agents have been suggested. There is significant evidence that mechanisms of vascular remodeling, needed to restore vessel integrity and
function after this initial injury, are abnormal in SSc. This adverse remodeling leads to pathologic intimal proliferation and medial hypertrophy, culminating in luminal narrowing (or obliteration) and tissue ischemia.
13 The abnormally thickened vessel walls promote intravascular thrombosis from platelet aggregation and further contribute to luminal narrowing. Angiogenesis and vasculogenesis also appear dysregulated in SSc, leading to vascular malformations and an impaired ability to generate new functional microvessels.
10Functional abnormalities of the vascular system are superimposed on structural abnormalities.
13 Vascular instability and altered vasoreactivity are prominent features in SSc and further compromise the vascular compartment.
10 An imbalance between vasoconstrictive and vasodilating mediators plays a role. Endothelin 1 (ET-1), released by injured endothelial cells, has received much attention in this regard. ET-1 is not only a potent vasoconstrictor but is also known to be profibrogenic, enhancing fibroblast proliferation and collagen synthesis. Thus, ET-1 may be an important link between the observed vasculopathy and pathologic fibrosis.
Tissue fibrosis dominates the second phase of this disease.
8 Persistent and uncontrolled upregulation of collagen gene expression by recruited fibroblasts and myofibroblasts leads to excessive deposition of collagen and other extracellular matrix (ECM) proteins. Complex autocrine and paracrine signaling loops sustain and amplify the abnormal fibrogenic response. Some of the relevant mediators in the signaling loops include the cytokines interleukin (IL)-1, IL-2, IL-4, and IL-6, activating factors such as transforming growth factor beta (TGF-β), platelet-derived growth factor and connective tissue growth factor, and monocyte chemotactic protein-1.
9 There is also increasing recognition that tissue hypoxia, which occurs in the context of the aforementioned vascular abnormalities, perpetuates the cycle of fibrosis and vascular pathology. Several mechanisms have been postulated: (1) hypoxia stimulates the production of extracellular matrix proteins via hypoxia-inducible factor-1 α-dependent and α-independent pathways; (2) hypoxia is also a potent inducer of vascular endothelial growth factor (VEGF), which when chronically overexpressed, leads to the formation of chaotic vessels with decreased blood flow.
14,
15 Thus, hypoxia may be another link between vasculopathy and fibrosis in SSc.
Although it is generally accepted that autoimmunity has a role in disease pathogenesis, many issues remain unresolved.
8,
12 It is unclear whether immune dysfunction is involved in initiation and/or disease maintenance. It is also not clear how the pathways of vascular pathology, fibrosis, and autoimmunity intersect. Numerous disturbances of both the humoral and cell-mediated immune systems have been described in different subsets of SSc patients at varying stages of the disease with different clinical manifestations.
9 Conflicting and inconsistent reports regarding these immune abnormalities (related in part to the heterogeneity of the populations studied) have led to difficulty with the interpretation of such findings and uncertainty regarding their relevance. Nevertheless, considerable evidence indicates that a skewed balance between type 1 and type 2 helper T cells toward Th2 activation is important for development of fibrosis. Emerging data also suggest that IL-17-producing T cells (Th17) may be relevant in pathogenesis.
9Numerous autoantibodies are also detected in the sera of SSc patients.
16 These include antinuclear antibodies such as antitopoisomerase 1 (anti-Scl 70) and anticentromere, as well as antinuclear antibodies (i.e., anti-RNA polymerase III and anti-U3-fibrillarin). Much is known about the associations of these antibodies with clinical subsets and patterns of organ involvement in SSc (i.e., anticentromere with limited SSc; anti-Scl 70 with diffuse SSs). However, to date, evidence that these autoantibodies are pathogenic has not been firmly established. More recently, several additional autoantibodies directed against nonnuclear antigens have been detected. These include antiendothelial cell antibodies, antifibrillin-1 antibodies, antimatrix metalloproteinase antibodies, and antiplatelet-derived growth factor receptor antibodies.
17 Experimental evidence suggests that these antibodies may be relevant in disease pathogenesis by initiating and/or propagating tissue damage. However, confirmatory studies are needed.
Scleroderma Renal Crisis
Scleroderma renal crisis (SRC) is one of the most devastating and life-threatening complications of systemic sclerosis. It develops in 5% to 10% of SSc patients and is seen almost exclusively in patients with diffuse systemic sclerosis. Two large cohort studies reviewed the clinical characteristics of SSc patients who develop this renal complication.
2,
18 Penn et al.
18 described the course of 1997 SSc patients seen at a single institution between 1990 and 2005 and reported that 110 (5.5%) developed SRC. Of these, 86 patients (78%) had diffuse disease and 24 (22%) had limited disease. These data are comparable to those of other studies.
Scleroderma renal crisis usually develops early in the course of the disease. Approximately two thirds of affected patients carry the diagnosis of SSc for less than 1 year and almost all SRC cases are diagnosed within 5 years of the onset of SSc. Patients typically present with a precipitous onset of severe hypertension and rapid deterioration of renal function with oliguria or anuria. This is often accompanied by signs of microangiopathic hemolysis. Clinical manifestations of SRC are mainly those of malignant hypertension (e.g., headache and/or seizures from hypertensive encephalopathy and visual disturbances from hypertensive retinopathy). Dyspnea may be due to acute left ventricular failure and pulmonary edema due to the effects of malignant hypertension on the myocardium and volume overload from oliguria. Associated laboratory findings include elevated serum creatinine that progressively and rapidly rises and may be accompanied by proteinuria, which is usually in the subnephrotic range (<2.5 g per 24 hours). Nephrotic range proteinuria is uncommon. Urine sediment may be normal or may reveal microscopic hematuria with few cells or casts but a nephritic sediment is unusual. Anemia, thrombocytopenia, and schistocytes in the peripheral blood smear support the presence of a microangiopathic hemolytic process along with other
markers of intravascular hemolysis such as elevated lactate dehydrogenase (LDH) and low haptoglobin.
Atypical presentations may make the syndrome more difficult to recognize, leading to missed diagnoses. Normotensive renal crisis occurs in 10% of cases of SRC.
19 Although blood pressures are normal or only modestly elevated in this variant of renal crisis, they are often higher than the patient’s baseline blood pressure. This subtle change can serve as an important clue to the diagnosis and underscores the need to closely monitor blood pressures in SSc patients (particularly those at a high risk of SRC). Thrombotic microangiopathy is often present in normotensive SRC and its presence should also raise suspicion of this syndrome.
20 Renal crisis can also occur rarely in a subset of patients who have no significant dermal sclerosis (referred to as sclerosis sine scleroderma). These patients have characteristic internal organ involvement but an absence of detectable skin features. Finally, in one quarter of patients, the diagnosis of SRC will precede a formal diagnosis of systemic sclerosis.
18,
21One of the challenges in diagnosing SRC, in both typical and atypical presentations, is distinguishing it from other forms of thrombotic microangiopathy (i.e., thrombotic thrombocytopenic purpura/hemolytic uremic syndrome [TTP/HUS]) given the similarities in presentation and laboratory abnormalities. This distinction is important as therapies differ. A renal biopsy does not reliably distinguish between these disorders. Rather, the clinical diagnosis relies on a compatible history, an evaluation of appropriate serologies, and a careful assessment of risk factors. Several factors may help to identify patients at a greater risk of developing SRC.
22 These include early diffuse systemic sclerosis, rapidly progressive skin thickening, new onset anemia, and new cardiac events (such as heart failure or pericardial effusion). Use of glucocorticoids (≥ 15 mg per day or the equivalent) in the preceding 6 months have long been considered a potential precipitant in SRC. In a case control study, Steen and Medsger
23 reported that high dose corticosteroids were administered more frequently in SRC patients (36%) than in controls (12%) (odds ratio [OR], 4.37). Although this association has also been reported by other investigators, causality is difficult to prove.
21 Rather, corticosteroids may have a confounding role because patients who are most likely to receive corticosteroids are also those at the highest risk for SRC. Nevertheless, the avoidance of corticosteroids in patients at risk is prudent. Autoantibody profiles may also provide clues regarding the risk of SRC. Anti-RNA polymerase III antibodies are associated with the development of SRC. Antifibrillarin or anti-U3-RNP antibodies may also identify patients at risk of developing internal organ manifestations, including SRC.
24 Conversely, patients with anticentromere antibodies are less likely to develop SRC.
18
Pathophysiology of Scleroderma Renal Crisis
Injury to vascular endothelial walls, which underlies the pathophysiology of SSc in general, is considered a primary event in SRC. The vascular insult in SRC occurs in the interlobular and arcuate arteries of the renal cortex. The resulting thickening and proliferation of the intima, the deposition of glycoproteins and mucopolysaccharides, and the formation of platelet microthrombi lead to luminal narrowing and reduced renal perfusion. Chronic renal cortical ischemia leads to hyperplasia of the juxtaglomerular apparatus, stimulation of renin release, and activation of the renin-angiotensin-aldosterone system (RAAS). Hyperreninemia and ongoing activation of the RAAS is well recognized as playing a pivotal role in SRC by perpetuating intrarenal vasoconstriction, exacerbating renal ischemia, and inducing hypertension.
25 However, RAAS activation alone appears insufficient to initiate the full expression of SRC. Indeed, investigators have found that plasma renin levels can be markedly elevated for some time prior to the onset of SRC and may even be seen in patients that do not develop SRC.
26 Thus, an additional trigger(s), superimposed on a system “primed” by renin excess, is believed necessary to set in motion the explosive cascade of events of SRC. This trigger is unknown, but it has been proposed that conditions or stimuli that acutely compromise renal perfusion such as dehydration, sepsis, cardiac dysfunction, or intense vasospasm of intrarenal arterioles (possibly from cold exposure or other stressors) may contribute.
26,
27,
28Dysregulation in the endothelin system has also been implicated in the pathophysiology of SRC. Levels of ET-1 are increased in SSc patients with renal crisis.
29,
30 One pathology study compared the distribution of ET-1 (by immunohistochemistry) in renal biopsies from SRC patients and patients with other vascular diseases involving the kidney.
31 Increased expression of both ET-1 and ET-1 receptors was detected in the small renal arteries of SRC patients, and the pattern of endothelial staining for ET-1 in both glomeruli and arteriolar lesions appeared specific for renal crisis.
The Role of a Renal Biopsy
A renal biopsy is usually not required to establish the diagnosis of SRC. Histologic changes are not pathognomonic for SRC and can be seen in other conditions that share endothelial injury as an underlying mechanism, such as malignant hypertension, TTP/HUS, and antiphospholipid syndrome. A renal biopsy is warranted when there is diagnostic uncertainty and/or to exclude other pathologies that warrant different management, such as crescentic glomerulonephritis or other inflammatory glomerular diseases.
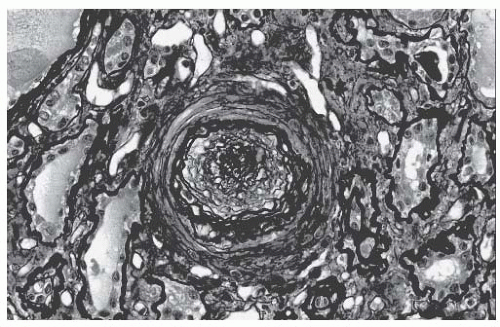
The pathologic changes of SRC are evident in the small interlobular and arcuate arteries.
32,
33 There is edema and thickening of the intima from mucin accumulation, and the characteristic onion skin lesion is due to concentrically arranged myointimal cellular proliferation and fibrosis, as shown in
Figure 54.1. Both contribute to a luminal occlusion. Thrombosis and fibrinoid necrosis may be seen. The juxtaglomerular apparatus may appear particularly prominent, which is consistent with hyperreninemia from a reduced renal perfusion. Glomerular changes are variable and may be related to renal ischemia or direct glomerular endothelial injury. There may be a collapse of capillary loops or a thickening of
capillary walls with a double contour appearance (on silver or periodic acid Schiff [PAS] staining), which are due to fibrin thrombi. Immune complexes are not present.
Treatment of Scleroderma Renal Crisis
Early diagnosis and prompt, aggressive treatment with ACE inhibitors (ACE-I) is crucial to prevent irreversible renal damage and a potentially fatal course. The effectiveness of ACE-I in halting progression and even reversing the process may be attributed to the interruption of the renin-angiotensin system, and AT-II-induced vasoconstriction. The angiotensin-converting enzyme also degrades bradykinin, a potent vasodilator. Interfering with this degradation of bradykinin may also contribute to the positive effects of ACE-I.
Most experience in SRC is with captopril, the agent used in early studies. There are fewer data available regarding the efficacy of other ACE-I in this setting, but they likely provide comparable benefit. Nevertheless, advantages of captopril in the acute setting include a rapid onset of action and a short duration of action, which provide more flexibility for dose titration. Captopril is usually initiated at 6.25 to 12.5 mg every 8 hours. The dose is titrated up (to 50 mg three times daily) to achieve gradual but steady blood pressure reduction. The goal is to return the patient to baseline blood pressures within 72 hours. Some recommend that blood pressure reductions should not exceed 20 mm Hg per 24 hours because this may compromise renal perfusion and should increase the risk of acute tubular necrosis. Even with judicious control of blood pressure, serum creatinine may rise with ACE-I therapy because of the associated decrease in efferent arteriolar resistance and intraglomerular pressure. However, this is not an indication for the discontinuation of therapy. Treatment with ACE-I is also indicated for patients with normotensive renal crisis, but escalation should be done carefully to avoid hypotension.
Data regarding the efficacy of AT-II receptor blockers (ARB) for the initial management of SRC are less clear. Some have reported benefit; others suggest suboptimal blood pressure control and greater rates of renal failure in patients with SRC treated with ARBs alone and subsequent improvement in renal function with the substitution of ACE-I.
34 The reason for such differences is unknown, but a possible explanation may be that ARBs, unlike ACE-I, do not inhibit the degradation of bradykinin.
If ACE inhibitors (at maximum recommended doses) are not sufficient to lower the blood pressure, other antihypertensive agents should be added. There are no studies that address which agents are most effective in this setting. Diuretics are best avoided (unless there is a strong clinical indication) because they may stimulate renin level. β-Blockers have the theoretic potential to worsen Raynaud phenomenon. Thus avoidance, if possible, might be prudent. Calcium channel blockers and vasodilators are reasonable options. Blocking the RAAS at multiple sites is an attractive approach in SRC. Aliskiren, a direct renin inhibitor, has theoretic benefit to further attenuate the RAAS in SRC and lower blood pressure. By blocking the catalytic activity of renin at the point of activation of the RAAS, aliskiren blocks the synthesis of all angiotensin peptides. This prevents the compensatory increase in renin that can be seen with ACE-I and ARB. At present, no studies have explored direct renin inhibition for therapy of SRC.
Despite the overall impressive impact of ACE-I on disease course, individual responses vary and many patients still suffer the full expression of SRC. The extent of this problem is highlighted in studies by Steen and Medsger
35 and Penn et al.,
18 which reviewed the course of SRC patients treated immediately (and aggressively) with ACE inhibitors. Permanent dialysis was required in 20% and 41% of these patients, respectively. This underscores the need for additional therapeutic strategies (beyond blockade of the RAAS) for this devastating complication. Intravenous prostacyclin and its analog, iloprost, have been used at some centers during the hypertensive phase of the renal crisis based on anecdotal observations of benefit.
36,
37 Prostacyclin mediates vasodilation and has been reported to increase renal perfusion. There are no formal trials addressing the role of prostanoids as adjunctive therapy to angiotensin converting enzyme [ACE-I] in SRC.
37 HMG-CoA reductase inhibitors (statins) have been proposed for the treatment of SRC and possible prophylaxis.
38 This is based on evidence suggesting that statins may have a direct protective effect on endothelium, in addition to their well-known cholesterol lowering effect. ET is also a potential target of interest. The use of ET receptor antagonists in SRC is a particularly compelling idea in light of the possible role of ET in this disorder. Apart from their vasodilating effects, ET receptor antagonists may also reduce the profibrotic effects of endothelin. Data regarding the use of ET-receptor antagonism in SRC are limited. In a small
pilot study, six patients with SRC were treated with bosentan, a nonselective ET-receptor antagonist, within 6 weeks of their diagnosis.
38a All patients were also receiving ACE-I at full therapeutic doses. The treatment regimen consisted of bosentan at 62.5 mg for 1 month and then 125 mg twice daily for 5 months. Bosentan was well tolerated. Overall, mortality and dialysis rates were not significantly different compared to a historic cohort receiving standard therapy. Rebound phenomena (i.e., Raynaud phenomenon, hypertension), occurred in half of the patients upon the withdrawal of therapy. A larger, open label trial using bosentan is currently recruiting in France to more fully assess efficacy of this drug in SRC. (clinical trials.gov: Effect of Bosentan in Scleroderma Renal Crisis/ScS-REINBO)
Prognosis
Prior to the availability and aggressive use of ACE inhibitors, the prognosis of SRC was abysmal with progression to end stage kidney disease (ESKD) over a period of 1 to 2 months and death usually within 1 year. The benefits of ACE-I in SRC were supported in an early single center study by Steen et al.,
39 which compared outcomes before and after the availability of ACE-I. One-year survival improved from 15% to 76% after the introduction of ACE-I therapy. Five-year survival improved from 10% to 65% with treatment. No randomized controlled trials have performed a head-tohead comparison of other therapies versus ACE-I and, given the evidence, there likely will never be.
In patients who do not require dialysis during their course of SRC, improvements in renal function are detectable for several years, suggesting that recovery is a slow, prolonged process that likely includes vascular remodeling.
18 Among patients who require renal replacement therapy during the acute episode of SRC, more than half may recover sufficient renal function to permanently discontinue dialysis within 12 to 18 months.
35 The continuation of ACE inhibitors is recommended during dialysis while monitoring for signs of renal recovery. Long-term survival of patients who never require dialysis or only need temporary dialysis seems to be comparable to patients with diffuse disease without a renal crisis. In contrast, long-term survival is less favorable for patients who remain dialysis dependent. These patients appear to have a higher mortality compared to those with end-stage kidney disease for other reasons.
It is noteworthy that the prognosis of patients with normotensive SRC appears worse than in hypertensive patients.
20 The basis for this is unclear, but hypotheses have been proposed. Difficulty in recognizing SRC in an atypical form may lead to a delay in diagnosis and management, allowing ongoing subclinical injury that may be irreversible. Alternatively, different pathogenetic mechanisms may underlie this form of SRC, which may be less dependent on the activation of the RAAS and thus, less responsive to ACE inhibitors. Other risk factors associated with a poor outcome in SRC include male sex, older age, the presence of congestive heart failure, serum creatinine levels greater than 3 mg per deciliter at the initiation of treatment, and a time period of more than 3 days to control blood pressure.
35,
39
Renal Transplant
A renal transplant is an acceptable option for those who progress to end-stage kidney disease and who fail to recover renal function due to SRC. Transplantation may offer a survival advantage compared to patients who remain on dialysis.
40 However, in light of the potential for delayed renal recovery, decisions regarding renal transplantation should be deferred for at least 2 years. Renal allograft outcomes are reasonable, although they may be reduced compared to the general renal transplant population.
40,
41Recurrence of SRC is a concern but is uncommon. It is estimated to occur in less than 5% of cases.
42 Most of the reported cases recurred relatively early in the posttransplant course within 2 years. Establishing the diagnosis of recurrent SRC in the allograft can be particularly challenging because other processes such as acute antibody mediation rejection, chronic transplant vasculopathy, and acute calcineurin inhibitor nephrotoxicity produce a similar histologic appearance. To reduce the risk of recurrent disease, high dose glucocorticoids should be used judiciously given their potential role in precipitating a renal crisis. When unavoidable, limiting the dose and duration is recommended. Calcineurin inhibitors have also been associated with precipitating renal crises in case reports. Thus, it may be prudent to consider alternative immunosuppressants.
Monitoring and Prevention
The unpredictable onset of SRC and the importance of a prompt diagnosis underscores the necessity of careful monitoring of SSc patients, particularly those considered at the highest risk for this complication. Educating patients about symptoms and signs of SRC is important, and consistent home blood pressure monitoring should be encouraged.
Prophylactic use of ACE-I or ARB do not appear to protect against the development of SRC.
21,
23 Retrospective and case control studies have found neither benefit nor harm with ACE inhibitors related to the development of SRC,
23 although some investigators have noted a trend toward worse renal outcomes.
18,
21 Further investigations are needed to clarify these findings.
Antineutrophil Cytoplasmic Antibody-Associated Renal Disease
Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is another distinct cause of rapidly progressive renal failure in SSc patients. The concurrence of AAV and SSc is a relatively rare complication described only in case reports and small cases series.
43,
44,
45,
46 Nevertheless, AAV is an important entity to consider in the differential diagnosis of acute kidney injury in SSc patients and needs to be distinguished from SRC, which may have a similar clinical presentation but a completely different therapeutic approach.
AAV can occur in both the diffuse and limited variants of SSc, and it has also been reported in patients with systemic sclerosis sine scleroderma.
47,
48 It is most common in the fifth and sixth decade of life and has a similar gender distribution as all SSC patients (female to male ratio, 4:1). Nearly all SSc patients with concurrent AAV have ANCAs with a perinuclear staining pattern (p-ANCA) directed against myeloperoxidase (MPO-ANCA). Clinical manifestations are more consistent with microscopic polyangiitis rather than granulomatosis with polyangiitis (Wegener granulomatosis). ANCAs directed against PR3 ANCA are rarely detected in this population.
49Certain presenting clinical and laboratory features may help to distinguish AAV from SRC (
Table 54.1).
44,
46 ANCA-associated kidney disease tends to occur later in the course of SSc, whereas SRC generally occurs earlier. The average duration of SSc prior to the onset of AAV and SRC is 7.8 years and 3.2 years, respectively. In contrast to SRC, which is associated with severe hypertension, blood pressure in ANCA-related acute kidney injury tends to be normal, although mild-to-moderate hypertension may be present in up to one third of patients. As such, AAV may be confused with and misdiagnosed as normotensive SRC. Patients with AAV in the context of SSc may present with other manifestations of vasculitis including limb ischemia, cutaneous lesions, and neuromuscular involvement (i.e., mononeuritis multiplex). The presence of such findings favors a diagnosis of AAV over SRC. Also, the vasculitis of AAV is an inflammatory vasculitis, whereas SSc is a noninflammatory vasculopathy. Thus, evidence of inflammation such as fever and elevated acute phase reactants (erythrocyte sedimentation rate [ESR], C-reactive protein [CRP]) favors a diagnosis of AAV rather than SRC. Pulmonary hemorrhage is a frequent vasculitic manifestation leading to respiratory distress in this setting (pulmonary renal syndrome). Distinguishing an alveolar hemorrhage from a pulmonary edema, which may occur in SRC, is often difficult because the symptoms and radiologic imaging may be similar. Anemia can be present in both, but is usually normochromic normocytic in AAV and microangiopathic in SRC. The urine sediment is active and nephritic in AAV but typically is bland in SRC.
If AAV is suspected, tests for ANCA and for anti-MPO and anti-PR3 by enzyme-linked immunosorbent assay (ELISA) are warranted, but results are usually not available in a timely manner. A kidney biopsy should be performed expeditiously
to confirm the diagnosis. A histology is consistent with pauci-immune necrotizing and crescentic glomerulonephritis.
The treatment of AAV requires aggressive immunosuppression consisting of induction with high dose corticosteroids and cyclophosphamide with or without plasma exchange, followed by maintenance immunosuppressive therapy. B-cell depletion with rituximab might also be considered. Although data are limited, there are no reported cases of steroids promoting the development of SRC during the treatment of AAV despite the theoretic risk. Because prognosis is so poor without therapy, intensive immunosuppression, including steroids, is recommended.
What predisposes some SSc patients to the development of a superimposed vasculitis is not known. In some cases, a hypersensitivity reaction to the medication, D-penicillamine, has been considered to be a trigger, but in the vast majority, a cause is not identified. ANCAs have also been detected (rarely) in the sera of unselected SSc patients during screening evaluations.
44,
46,
49,
50,
51 The significance of ANCA in SSc patients without clinically evident vasculitis is controversial. Some authors suggest that this finding is considered a red flag, indicating an inflammatory component to the underlying disease, whereas others have found no increased risk of renal disease or other vasculitic complications.
50
Other Renal Manifestations in Systemic Sclerosis
Other renal abnormalities may occur in SSc patients that are not as overt or as dramatic as SRC or ANCA vasculitis. Estimates of kidney involvement in SSc vary widely based on different definitions of kidney injury and the markers of renal disease that are examined.
52,
53,
54,
55,
56,
57,
58 One report identified abnormal renal function (defined as creatinine ≥1.2 mg per deciliter) or proteinuria (defined as 3 or 4+ proteinuria on dipstick on two occasions) in 16% and 13%, respectively, of patients with dcSSc (without a history of SRC).
57 Over a mean follow-up of 10 years, only 2 of 546 patients in this cohort reached end-stage kidney disease, suggesting a benign clinical course for the vast majority. A greater percentage of patients are considered to be affected if more sensitive measures of GFR are used.
53 One study measured GFR using technetium 99mDTPA in 31 patients with normal serum creatinine and showed a reduction of GFR in 55% of patients, with 32% categorized as stage II chronic kidney disease (CKD) (60 to 89 mL per minute) and 23% as stage III (30 to 59 mL per minute).
55 When pathologic renal change at an autopsy is used as the criterion, approximately 60% of patients will have histologic evidence of kidney involvement, indicating that subclinical disease is relatively common in SSc.
52,
58Renal involvement in SSc patients may be related or unrelated to the underlying disease process, other organ involvement (i.e., heart failure, pulmonary hypertension), or associated therapies (
Table 54.2). Treatment with D-penicillamine, which had been used for years for the management of skin involvement in dcSSc, has been associated with renal injury and proteinuria.
59 Up to 20% of treated patients develop membranous nephropathy, which resolves with drug cessation.
60 Diffuse proliferative glomerulonephritis, pulmonary renal syndrome, drug-induced lupus syndrome, and ANCA-related vasculitis have also been reported to be associated with D-penicillamine treatment.
61,
62 Due to its high rate of side effects, including renal toxicity and questionable therapeutic effect, penicillamine is now rarely used in the treatment of SSc.
An important association exists between the presence of pulmonary hypertension and a reduced glomerular filtration rate. Campo et al.
63 evaluated 76 SSc patients with pulmonary arterial hypertension and reported that 45.6% had renal dysfunction (estimated glomerular filtration rate [eGFR] <60 mL/min/1.73 m
2) at the time of diagnosis.
63 Only 6.5% of these patients had a prior episode of renal crisis. eGFR was a strong predictor of survival in this cohort, with an eGFR less than 60 mL/min/1.73 m
2 associated with a threefold risk of mortality. The reduction in GFR may reflect simultaneous structural damage to both the pulmonary and the renal vascular beds, or may be related to the prerenal effects of severe pulmonary hypertension.
Other studies have identified proteinuria as an independent risk factor for death in SSc patients with a hazard ratio of 3.34.
64,
65,
66 Although the mechanism for this association is not clear, it has been proposed that the presence of proteinuria may be a marker of more severe underlying endothelial dysfunction, which could portend a worse prognosis.
Novel Therapies for Systemic Sclerosis and the Potential for Renal Injury
Nonselective immunosuppressive medications (i.e., cyclophosphamide, azathioprine, prednisone, methotrexate) are frequently used to treat the complications of SSc despite a
lack of convincing data that these therapies reverse the natural disease progression.
67 Therapeutic attempts at selectively blocking fibrotic pathways have not met with much success, probably because of the complexity of the fibrotic process with its multiple layers of regulation. A number of newer biologic agents targeting other molecular pathways, cellular effectors, and signaling molecules believed to be operative in SSc are now available and are under clinical investigation.
68Autologous hematopoietic stem cell transplantation (HSCT) has also been applied for selected SSc patients with a poor prognosis (predominantly with diffuse cutaneous disease and severe internal organ involvement).
69 The rationale for HSCT in SSc is to reset the dysregulated immune system. Phase I/II trials of HSCT have demonstrated a reversal of skin fibrosis, improved functionality and quality of life, and stabilization of the internal organ function in SSc patients.
70,
71,
72,
73,
74 Experience is still limited and toxicity remains a concern. The potential for renal complications in SSc patients deserves special consideration.
Acute kidney injury (AKI) is not an uncommon complication after HSCT irrespective of the transplant indication. Kidney injury may manifest as thrombotic microangiopathy, radiation nephritis, glomerular disease, and acute tubular necrosis (ATN) (among others) and may be attributed to such factors as conditioning regimens, total body irradiation (TBI), nephrotoxic drugs (i.e., calcineurin inhibitors), and infections. Whether having SSc, with the associated underlying vasculopathy, conveys a higher risk of kidney injury after HSCT, particularly if exposed to radiation (which has effects on vascular endothelium), glucocorticoids, or calcineurin inhibitors, is unclear. Furthermore, both TMA and radiation nephritis can clinically mimic SRC. Distinguishing among these causes of AKI can be particularly challenging in SSc patients after HSCT. Until further data are available, measures to minimize nephrotoxicity will be important. These include aggressive control of blood pressure, the restricted use of glucocorticoids and calcineurin inhibitors, the avoidance of TBI or renal shielding, and close monitoring of renal function.
Presently, there are three ongoing randomized trials investigating the safety and efficacy of autologous HSCT for SSc: Autologous Stem Cell Transplantation International Scleroderma (ASTIS), American Systemic Sclerosis Immune Suppression Versus Transplant (ASSIST), and Scleroderma Cyclophosphamide Versus Transplant (SCOT). ASSIST and ASTIS use a nonmyeloablative regimen and SCOT uses a myeloablative regimen with total body irradiation. Hopefully, results from these trials will clarify the role of HSCT in SSc and will address the risk of kidney injury in these patients.
RHEUMATOID ARTHRITIS
Rheumatoid arthritis (RA) is an inflammatory polyarthritis with a peak age of onset between 40 and 60 years and with a greater than twofold increased prevalence in women. RA is generally considered to represent an autoimmune disorder because of its characteristic laboratory profile of autoantibodies to cyclic citrullinated peptides (anti-CCP), to immunoglobulin G (rheumatoid factor), and to nuclear antigens (antinuclear antibodies [ANA]). There are numerous defects in cytokine production and cell-mediated immunoregulatory pathways that are integral components of the pathogenic inflammation and autoreactivity. Some investigators hypothesize, based on animal models and on epidemiologic evidence, that certain infections can be the triggering event, which in a susceptible person with genetically determined or otherwise acquired defects in immunoregulatory circuits leads to RA.
75
Clinical Features of Rheumatoid Arthritis
RA is characterized by symmetrical stiffness and painful swellings (inflammation and effusions), typically of multiple joints of the upper and lower extremities; if these findings persist for more than 6 weeks, they fulfill a key diagnostic criterion for RA. Because RA typically progresses to a chronic disease, persistent or recurring synovitis leads to joint effusions and the destruction of cartilage and erosions of periarticular bone, eventuating in deforming arthropathies. Fatigability, malaise, anorexia, and weight loss are common debilitating features of RA, reflecting high circulating levels of immune complexes, acute phase reactants, and cytokines. The protracted inflammatory state, when not interdicted by effective disease remitting treatment, appears to be a major contributor to accelerated atherosclerotic cardiovascular disease, which in turn leads to increased rates of debilitating morbidity and premature mortality in patients with RA.
76
General Treatment Strategies for Rheumatoid Arthritis
Multiple studies have shown definitively that destructive pathologic processes start very early in the course of RA, and that there are long-term benefits of aggressive therapy on the natural history of RA. In the acute stage of the disease, the clinical and laboratory manifestations of RA typically respond dramatically to corticosteroids. However, because it is unusual to find a nontoxic dose of corticosteroid that can provide sustained control of RA, alternative therapies have been intensely pursued. Historically, there has been a succession of agents used adjunctively with corticosteroids for the treatment of RA, including general anti-inflammatory drugs (e.g., salicylates, nonsteroidal anti-inflammatory drugs [NSAIDs], antimalarials, gold salts, penicillamine, sulfasalazine), as well as more potent and directly immunosuppressive agents (e.g., methotrexate, cyclophosphamide, azathioprine, leflunomide). These agents, categorized as disease-modifying antirheumatic drugs (DMARDs), have shown objective salutary effects on clinical manifestations and long-term complications of RA; however, none of these agents predictably or consistently induces complete remissions and most have had excessive adverse side effects when used as maintenance therapy. For example, gold salts and penicillamine have been largely abandoned as treatment for RA due to suboptimal efficacy and their propensity to cause secondary membranous nephropathy.
Weekly doses of oral methotrexate have been the stalwart therapy for long-term management of RA for the past 3 decades. Methotrexate and low dose corticosteroid combination therapy produces satisfactory remissions in a substantial majority of cases. However, for patients with refractory or relapsing disease, biologic agents that antagonize tumor necrosis factor (e.g., etanercept, infliximab, adalimumab) are added to the basal methotrexate regimen. Other agents under active investigation as adjuncts to methotrexate include rituximab, anti-IL6-receptor (tocilizumab), and the costimulation inhibitor, CTLA4-Ig.
77,
78
Kidney Involvement in Rheumatoid Arthritis
Abnormalities of kidney function (e.g., abnormal urinalysis, proteinuria, diminished glomerular filtration rate [GFR], abnormal pathology) were recognized several decades ago in up to half of patients with RA. These clinical laboratory abnormalities were attributed to poorly controlled RA (e.g., secondary amyloidosis) or to the complications of the limited options for symptomatic treatment of RA (e.g., interstitial nephritis due to the protracted use of high dose aspirin).
79,
80 With subsequent availability of disease-modifying therapeutic strategies that provided more effective management of RA, the frequency of renal abnormalities has dramatically decreased and types of clinically significant renal complications have substantively changed over the past 2 to 3 decades. For example, both drug-induced interstitial nephritis and secondary amyloidosis have become increasingly rare due to the more effective treatment of RA. On the other hand, some types of glomerulonephritis do occur sporadically during the course of RA. The impacts on renal and patient survival of the different forms of glomerular disease vary from minor, in patients with isolated hematuria and low grade pathology, to major, in those with complicated nephritic and nephrotic syndromes and high grade pathology.
81