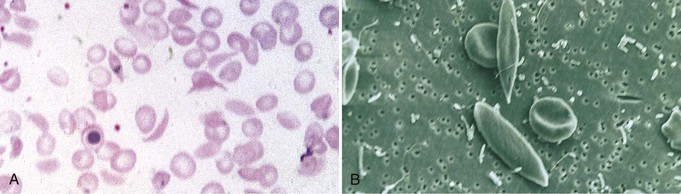
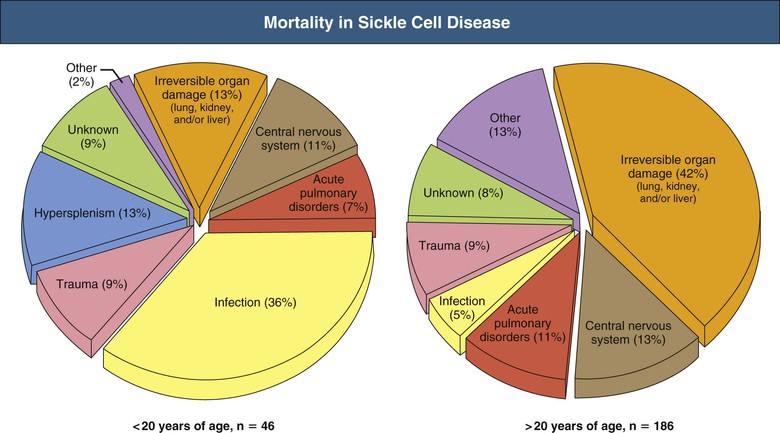
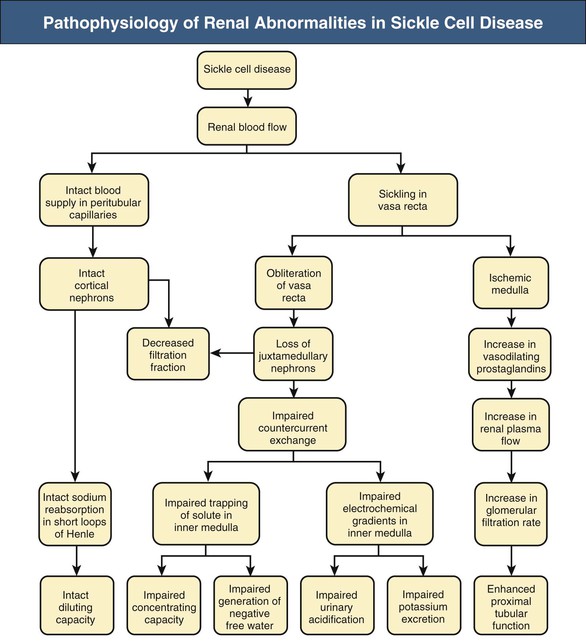
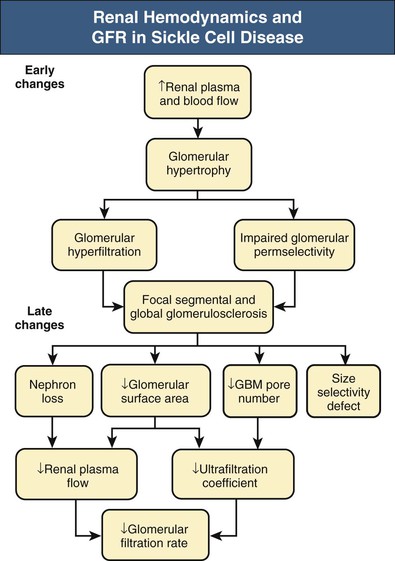
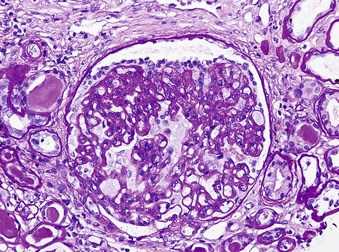
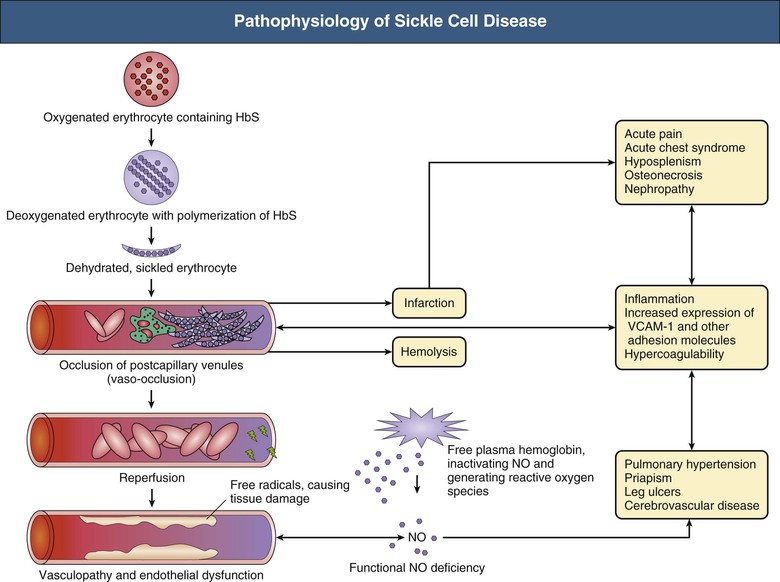
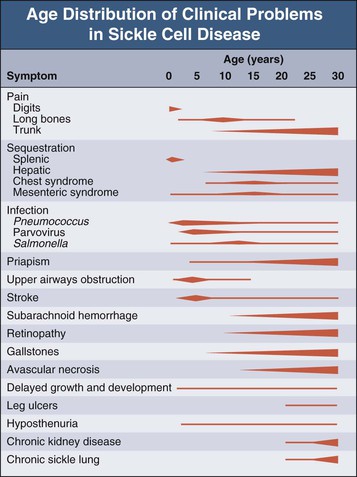
Jan C. ter Maaten, Fatiu A. Arogundade Sickle cell disease is an autosomal recessive inherited disorder that predominantly affects persons of African, Mediterranean, Indian, and Middle Eastern descent. The gene for sickle hemoglobin (hemoglobin S, or HbS) results in the replacement of the normal glutamine by valine in the sixth position of the β-globin subunit, thereby changing the configuration of the hemoglobin molecule and enhancing the aggregation of hemoglobin molecules during cellular or tissue hypoxia, dehydration, or oxidative stress. This aggregation decreases the pliability of the erythrocytes and may distort their shape to a characteristic crescent or sickle, resulting in their premature destruction (hemolysis) and frequent, widespread vaso-occlusive episodes with subsequent acute and chronic organ damage. Sickle cell anemia occurs in those homozygous for HbS or in heterozygotes when HbS coexists with another abnormal hemoglobin (e.g., hemoglobin C, β-thalassaemia chains). Sickle cell trait occurs in those heterozygous for HbS when the other hemoglobin molecule is normal. Sickle cell nephropathy describes the structural and functional abnormalities of the kidney in sickle cell disease. Sickle cell disease was first recognized in West Africa. The high prevalence of HbS in this region probably represents a survival benefit because the presence of sickle cell trait protects against malaria. At present, sickle cell disease is a worldwide health problem because HbS has spread throughout Africa, around the Mediterranean, and to the Middle East and India as well as to the Caribbean, North America, and northern Europe. The prevalence of the sickle cell gene is about 8% in African Americans and about 25% in adult Nigerians and in some areas in equatorial Africa, Saudi Arabia, and India. Restriction enzyme techniques have identified several HbS haplotypes, mutations of the HbS molecule, that have probably arisen independently of each other. There are four major types in Africa—the Benin, Senegal, Cameroon, and Bantu (or Central African Republic)—and one Asian haplotype. Variations in these haplotypes determine disease severity; for example, the Senegalese haplotype is associated with a higher hemoglobin F (HbF) concentration and has a better prognosis than others. In a sample population of Nigerians, Benin haplotype was detected in 92.3%.1 Gender influences disease severity; female HbSS patients with Benin haplotype have a higher HbF level than male patients. Sickle cell disease comprises a group of heterogeneous disorders that share the presence of the gene for HbS, either homozygous (i.e., sickle cell anemia, HbSS) or double heterozygous (i.e., combination of HbS with another abnormal hemoglobin).2,3 Sickle cell anemia is the most common form. The most common double heterozygous disorders are the combinations of HbS with hemoglobin C (HbSC) and with β-thalassemia (HbS-thal). Patients with HbS-thal may produce reduced amounts of normal β chains (HbS–β+-thal), but not always (HbS–β0-thal). Those with a sickle cell trait or carrier state are heterozygous for HbS only. The driving pathophysiologic factor is HbS polymerization. The mutated β-globin chains of the HbS molecule tend to crystallize to a polymer nucleus in the deoxygenated state. Polymerization disrupts the architecture and changes the shape of the red blood cell (RBC), increases its rigidity, and causes sickling (Fig. 51-1). Polymerization is dynamic and depends on three independent variables: degree of cellular hypoxia, intracellular hemoglobin concentration, and presence or absence of HbF (fetal hemoglobin).4 The main determinant of disease severity is the rate and extent of HbS polymerization, which drives the two major pathophysiologic processes: vaso-occlusion with ischemia-reperfusion injury and hemolytic anemia (Fig. 51-2).5 Vaso-occlusion occurs in all patients, often triggered by inflammation and typically found in clinical states of infection, hypoxia, hypovolemia, hypothermia, acidosis, and hyperosmolality. Vaso-occlusion is probably caused by dynamic endothelial-leukocyte-erythrocyte adhesive interactions in the postcapillary venules and precapillary obstruction by rigid, deformed erythrocytes. Episodic microvascular occlusion and ischemia can be interrupted by restoration of blood flow and reperfusion, which further promote inflammatory stress and tissue injury. Hemolysis has recently been shown to contribute to the development of progressive vasculopathy, characterized by endothelial dysfunction and proliferative changes in the intima and smooth muscle of blood vessels. The degree of hemolysis is associated with the development of pulmonary hypertension in sickle cell disease. An important role is ascribed to free hemoglobin, which inactivates nitric oxide and generates reactive oxygen species. Both these factors may further contribute to the hypercoagulability and vasculopathy. The clinical manifestations of sickle cell disease are individualized and age dependent (Fig. 51-3). A chronic low-grade hemolytic anemia always occurs and predisposes to gallstones. The most prevalent clinical problem is periodic crises of bone pain. During the first years of life, bone pain presents as the hand-foot syndrome, and over the years it can result in avascular necrosis of the heads of the femur and humerus. Other disabling complications include cerebrovascular accident (stroke) resulting from occlusion of major cerebral vessels, the acute chest syndrome, priapism, chronic leg ulceration, chronic pulmonary disease with pulmonary hypertension, and chronic kidney disease.6,7 Patients with sickle cell disease are prone to infections because of functional asplenia early in life as a result of splenic sequestration, recurrent splenic infarction, and consequent autosplenectomy. Common bacterial infections, especially with encapsulated organisms, can be fatal in these patients. Bacterial isolates during invasive bacterial infections show Streptococcus pneumoniae (38%), Salmonella spp. (33%), Haemophilus influenzae (14%), Escherichia coli (11%), and Klebsiella spp. (4%).8 S. pneumoniae and H. influenzae occur predominantly before 5 years of age, Salmonella increases almost linearly with age, and Klebsiella and E. coli predominate in patients older than 10 years. Pneumococcal infections carry high morbidity and mortality rates in the early years of life, necessitating vaccination or prophylaxis.2 Fever is common in patients with sickle cell disease. Common complications accompanying fever are painful crisis and acute chest syndrome. Bacteremia is not always confirmed, and in some cases, the fever may be viral in origin or related to infections from atypical organisms. Nevertheless, early antibiotic treatment is recommended pending microbiologic information. There are remarkable differences in clinical severity and outcome of disease. Sickle cell trait is a rather benign condition. Patients with HbSS tend to have more severe disease than those with HbSC. Likewise, patients with HbS–β+-thal do better than those with HbS–β0-thal. The Bantu haplotype is associated with the highest frequency of organ damage. However, the severity of disease may also differ among patients with an identical genotype. In part, these differences can be explained by the amount of HbF present because it protects against clinical severity. In addition, endothelial factors probably play an important role. Degree of adherence between sickle erythrocytes and endothelial cells has been shown to correlate with the clinical severity of disease. Also, an association has been found between circulating activated endothelial cells of microvascular origin and the onset of painful sickle cell crises.9 Life expectancy is reduced in sickle cell disease, especially in patients with symptomatic disease. An increased risk for early death is associated with low levels of HbF, renal failure, the acute chest syndrome, and seizures.7,10 Childhood survival to age 20 years has improved during the past decades. In the course of life, irreversible organ damage caused by arterial and capillary microcirculation obstructive vasculopathy gradually becomes more prevalent. The diagnosis of a clinically evident form of organ damage, such as leg ulcer, osteonecrosis, or retinopathy, predicts the development of a more lethal form of organ damage, such as chronic lung disease, renal failure, or stroke.11 Overall, the primary causes of death in patients with sickle cell anemia are chronic lung disease in 20%, renal failure in 14%, and stroke in 10%. In younger patients, the primary cause of death is infection; in older patients, the primary cause of death is irreversible organ damage (Fig. 51-4). Management of sickle cell disease is primarily directed at the relief of symptoms and prevention of complications, whereas newer treatments are being devised that target the pathophysiologic changes of the disease.12 Daily oral penicillin for children 2 to 5 years of age is effective in reducing both the infection rate and the mortality related to pneumococcal infection.13 Immunization against pneumococcus is recommended for children at 2 years of age, with booster doses at 5 years, although protection from current vaccines is imperfect. Immunization against influenza in children is also important. Empiric broad-spectrum antibiotics effective against gram-positive and gram-negative organisms should be used in adults with fever pending microbiologic confirmation. A common antibiotic is amoxicillin. Sickle cell crises are managed with oxygen therapy, rehydration with intravenous fluids, RBC transfusions, and analgesia. In severe cases, exchange RBC transfusions may be effective. Hydroxyurea treatment in patients with sickle cell anemia and recurrent vaso-occlusive events decreases the incidence of painful crises and acute chest syndrome and overall mortality. Butyrate compounds, deoxyazacytidine, and various combinations with erythropoietin are still being tried.12,14 Hematopoietic stem cell transplantation is potentially curative in sickle cell patients, associated with 80% to 85% disease-free survival in several series.15 Young and presymptomatic patients with high-risk features of severe disease benefit the most from hematopoietic stem cell transplantation. The hallmark of sickle cell nephropathy is the combination of impaired renal concentrating capacity and a normal diluting capacity.16,17 The defect in concentrating capacity results from loss of the countercurrent exchange mechanism in the inner renal medulla through loss of the vasa recta and long loops of Henle of the juxtamedullary nephrons (Fig. 51-5). The vasa recta of the juxtamedullary nephrons present an ideal setting for sickling. The renal medulla is relatively hypoxic and hyperosmotic, with increased blood viscosity in the medullary circulation and slow medullary blood flow. Studies in transgenic sickle cell mice have demonstrated distention and congestion of the vasa recta under hypoxic conditions. This environment facilitates the sickling of erythrocytes, formation of intravascular microthrombi, and obstruction of blood flow through the vasa recta. The loss of vasa recta has been confirmed by microradioangiographic studies18 (Fig. 51-6). Histologic examination of the medulla shows edema, focal scarring, and interstitial fibrosis resulting in tubular atrophy. Ischemic infarction in the vasa recta sometimes causes papillary necrosis. The concentration defect is found to be reversible in young children when sickling is prevented after multiple transfusions of normal blood, but it becomes irreversible thereafter. Adults with sickle cell anemia cannot concentrate the urine above 450 mOsm/kg H2O. This relates to the interstitial osmolality at the transition of the outer and inner medulla, at the tips of the short loops of Henle of the cortical nephrons. Patients with sickle cell trait or hybrid sickling disorders show intermediate concentrating defects. Maximal osmolality varies from 400 to 900 mOsm/kg H2O in sickle cell trait and from 400 to 700 mOsm/kg H2O in HbSC and declines further with aging. The diluting capacity is normal as a result of the intact reabsorptive function of the superficial loops of Henle of the cortical nephrons. These are supplied by peritubular capillaries, which present a less ideal setting for sickling than the vasa recta. In contrast to the diluting capacity, the free water reabsorption, or capacity to generate negative free water balance, is impaired by defective trapping of solute in the inner medulla. Urine volumes are typically higher than normal, a consequence of impaired concentrating ability. Response to a water deprivation test in sickle cell disease is usually positive with marked elevation of endogenous antidiuretic hormone, and response to desmopressin is poor. Because isosthenuria is typical, there is a susceptibility to normonatremic dehydration. Defects in urinary acidification and potassium excretion are other distal nephron function abnormalities.17 These abnormalities may become overt only when there is an increased supply of acid and potassium, as during rhabdomyolysis. The exact causes are unknown, but these defects probably reflect failure to maintain the necessary energy-requiring hydrogen ion (H+) and electrochemical gradients along the collecting ducts from the impaired medullary blood flow and hypoxia. The impaired potassium excretion is aldosterone independent. In contrast to the functional abnormalities of the distal nephron, proximal tubular function is enhanced. Reabsorption of phosphate and β2-microglobulin and secretion of uric acid and creatinine in the proximal tubule are increased. Therefore, creatinine clearance overestimates glomerular filtration rate (GFR) considerably. The cause of the enhanced proximal function is not clear, but it probably represents a secondary compensatory mechanism to correct for defects in medullary function. Renal hemodynamics show remarkable changes in the course of disease (Fig. 51-7). Young patients with sickle cell disease have increases in renal plasma flow (RPF) and renal blood flow and, to a lesser extent, in GFR. The increased RPF is ascribed to increased release of vasodilator prostaglandins as a result of medullary ischemia because prostaglandin inhibition significantly reduces RPF and GFR. It has been suggested that endothelial dysfunction from hemolysis can induce vasodilation in the cortex, including hyperfiltration and glomerular hypertrophy, in contrast to the prevailing vaso-occlusion in the medulla.19 The decrease in filtration fraction may be caused by selective damage of juxtamedullary nephrons, which have the highest filtration fractions. Glomerular hypertrophy is an early manifestation of sickle cell nephropathy. Histologic examination of the kidneys of young children shows glomerular enlargement and congestion, especially in the juxtamedullary glomeruli. Both afferent and efferent arterioles of these glomeruli may be dilated. Young adult patients with sickle cell anemia show a distinct pattern of glomerular dysfunction, with impaired glomerular permselectivity, increased ultrafiltration coefficient, glomerular hyperfiltration, and proteinuria.20,21 Prolonged glomerular hyperfiltration may cause further glomerular injury. This is supported by the common histologic finding of focal segmental glomerulosclerosis (FSGS) in adult patients with sickle cell disease22 (Fig. 51-8). Besides FSGS, other histopathologic variants are membranoproliferative glomerulonephritis, thrombotic microangiopathy, and specific sickle cell nephropathy.23 Hypoxia did not seem to play a key role in the progression of glomerular lesions in histopathologic studies because immunohistochemistry did not reveal a specific induction of hypoxic markers at the time of renal biopsy. In older patients, progressive ischemia and fibrosis with obliteration of glomeruli can be found. Glomerular studies show a decrease in glomerular basement membrane pore number and a size selectivity defect in patients with renal impairment.24 Good correlation exists between tubular and glomerular dysfunction, with both contributing to nephropathy in patients with sickle cell disease.25 Eventually, a combined decrease in ultrafiltration capacity and RPF can result in end-stage renal disease (ESRD).20
Sickle Cell Disease
Definitions
Sickle Cell Disease
Epidemiology
Pathogenesis
Genetics
Pathophysiology
Clinical Manifestations
Natural History
Treatment
Pathogenesis of Sickle Cell Nephropathy
Concentrating Capacity

Diluting Capacity
Other Tubular Abnormalities
Renal Hemodynamics
Glomerular Injury
Hormones in Sickle Cell Nephropathy
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Sickle Cell Disease
Chapter 51