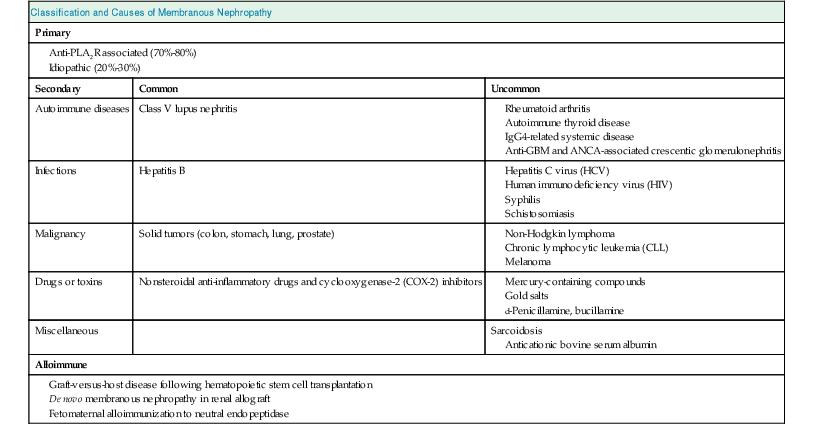
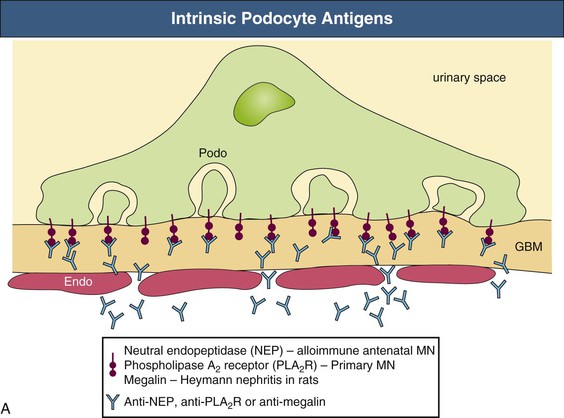
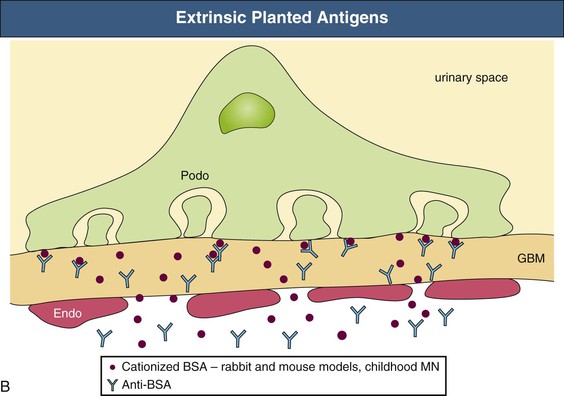
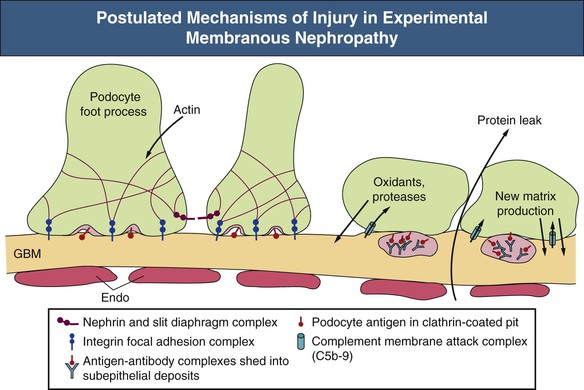
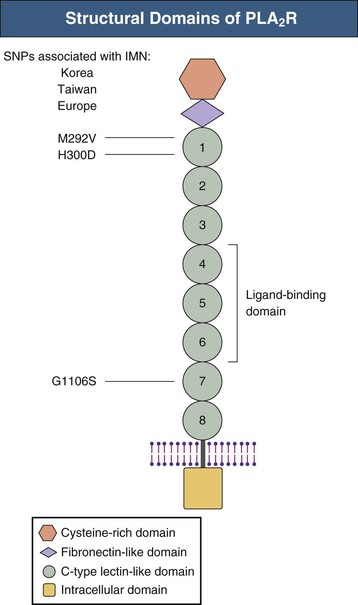
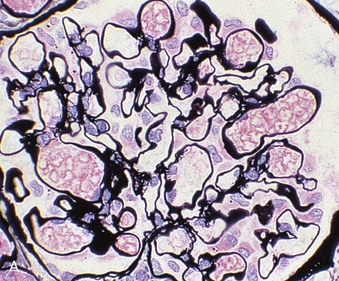
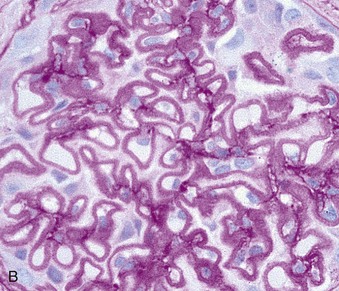
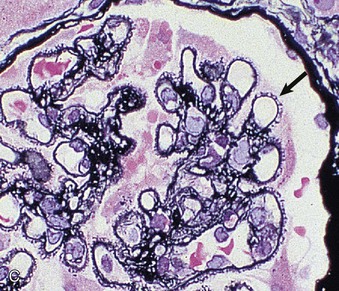
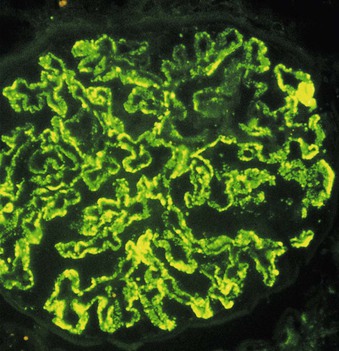
David J. Salant, Daniel C. Cattran Membranous nephropathy (MN) is an immune complex glomerular disease in which immune deposits of IgG and complement components develop predominantly or exclusively beneath podocytes on the subepithelial surface of the glomerular capillary wall. Podocyte injury resulting from the immune deposits increases glomerular permeability to plasma proteins, which results in proteinuria and potentially, nephrotic syndrome.1,2 In about 75% to 80% of patients, MN occurs in the absence of any identifiable cause or initiating event and is known as idiopathic or primary MN. Various conditions have been identified in association with MN, some of which are likely to be causal and some of which probably represent only associations1 (Table 20-1). Primary MN is an organ-specific autoimmune disease. It is the most common cause of primary nephrotic syndrome in older (>60 years) Caucasian adults, but the age range is broad, and patients may present for the first time in their teenage years.3 The term membranous refers to thickening of the glomerular capillary wall on light microscopy of a renal biopsy, but the entity now referred to as membranous nephropathy is defined by immunofluorescence and electron microscopy. These techniques reveal diffuse, finely granular immune deposits on immunofluorescence and electron-dense deposits in the subepithelial space that are now regarded as pathognomonic of MN. Consequently, MN is a pathologic diagnosis made in patients with proteinuria whose glomeruli exhibit these immune deposits without associated hypercellularity or inflammatory changes.2 Much of what we know about the pathogenesis of MN derives from observations in animal models.4,5 Studies of the Heymann nephritis model of MN in rats in the late 1970s established that the subepithelial immune deposits form in situ when circulating antibodies bind to an intrinsic antigen in the glomerular capillary wall. The antigen was subsequently identified as megalin, a large (~600 kD) transmembrane receptor of the low-density lipoprotein receptor family expressed on the basal surface of rat podocytes.6 Binding of circulating antimegalin antibodies induces capping and shedding of the antigen-antibody complexes, where they bind to the underlying glomerular basement membrane (GBM), resist degradation, and persist for weeks or months as immune deposits characteristic of MN (Fig. 20-1, A). Proteinuria in this model is caused by complement-fixing antibodies in the immune deposits that overcome local complement regulatory mechanisms and activate complement in situ. The primary mechanism involves sublethal podocyte injury induced by the complement membrane attack complex C5b-9, which triggers a cascade of structural and functional changes, including oxidative injury, calcium influx, activation of cytosolic phospholipase A2, production of arachidonic acid metabolites and cytokines, endoplasmic reticulum stress, DNA damage, and disruption of the actin cytoskeleton7 (Fig. 20-2). Podocyte foot process effacement is likely the result of the collapse of the actin cytoskeleton and loss of cell-GBM adhesion complexes, and the loss and displacement of slit diaphragms is associated with the onset of severe, nonselective proteinuria. Podocyte injury is also responsible for the production of new extracellular matrix (ECM) proteins that are laid down between and around the immune deposits, giving rise to the characteristic “spikes” and GBM thickening that are hallmarks of MN. Another mechanism of subepithelial immune deposit formation involves planted antigens5 (Fig. 20-1, B). This is best exemplified by animal models immunized with cationized bovine serum albumin (cBSA). The cBSA binds to negatively charged residues in the GBM, where it serves as a target for circulating anti-BSA antibodies. As in the Heymann nephritis model, podocyte injury and proteinuria result from local complement activation. Evidence has recently been obtained that both fixed and planted antigen mechanisms are involved in human MN (see Fig. 20-1). The first demonstration that circulating antibodies reactive with an intrinsic podocyte antigen may be involved in MN was provided by an unusual case of antenatal MN induced by the transplacental passage of alloantibodies to neutral endopeptidase (NEP), a known podocyte protein.8 The mother of the affected child was found to be deficient in NEP and had been immunized during a previous pregnancy and, like subsequent cases, produced complement-fixing anti-NEP alloantibodies. Although antibodies to anti-NEP do not account for primary MN, alloantibodies probably explain the development of de novo MN after renal transplantation and MN in the setting of chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation (see Table 20-1). The predominant autoimmune system responsible for primary MN is that associated with autoantibodies directed at the M-type phospholipase A2 receptor (PLA2R) on podocytes.9 Circulating anti-PLA2R antibodies are detectable in the serum of 75% to 80% of patients with primary MN from all ethnic groups and are rarely found in secondary MN.10,11 The antibodies are predominantly IgG4 and, as in the Heymann nephritis model, the antigen (PLA2R) and antibodies (anti-PLA2R) co-localize in the immune deposits in primary (but not in secondary) MN.9,12,13 PLA2R is a transmembrane protein of the mannose receptor family14 (Fig. 20-3). Although it has been shown to undergo constitutive endocytosis and is involved in the production of eicosanoids, reactive oxygen species, DNA damage, and cellular senescence, its role in podocytes is unknown. Reactivity to certain intracellular antigens, including aldose reductase, SOD2, and enolase, has been detected in primary MN and may contribute to the progression of podocyte injury.15 The best evidence of a planted antigen mechanism in MN is that described in a subset of children with MN who have been exposed to cBSA, presumably in bottled milk. In such cases, as in the animal models, the cBSA localizes in the GBM, where it forms complexes with circulating anti-BSA.16 Planted antigens may also be responsible for immune deposition in class V (membranous) lupus nephritis and hepatitis B virus (HBV)–associated MN. The resolution of MN depends on remission of the immune response, the extent of podocyte damage, and expansion of the GBM. In cases in which immunologic remission occurs before extensive podocyte loss and GBM remodeling, complete recovery is possible. On the other hand, proteinuria may persist for several weeks or months until the normal architecture is restored. Once there is extensive podocyte loss, severe proteinuria persists despite immunologic remission and glomerular sclerosis, tubular atrophy, and interstitial fibrosis may ensue. Membranous nephropathy may occur at any age and in all ethnic groups, but primary MN is more common in men than in women (2 : 1) and is rare in children. Primary MN has its peak incidence during the fourth and fifth decades of life (Box 20-1). In comparison, MN in childhood is more often secondary (e.g., caused by HBV). Primary MN is the most common cause of nephrotic syndrome in nondiabetic Caucasian adults, with an estimated annual incidence of 8 to 10 cases per 1 million population in Western countries. Although association with certain human leukocyte antigen (HLA) class II immune response genes indicated a genetic predisposition,17–21 primary MN is not a familial disease, except in rare patients with more than one affected family member.22 The genetic predisposition became more evident after the discovery of PLA2R as the major antigen, when studies from Korea and Taiwan documented a significant association with non-synonymous single-nucleotide polymorphisms (SNPs)23,24 in the first C-type lectin-like domain (CTLD) of PLA2R (Fig. 20-3), a region that is known to undergo conformational changes, as well as another in CTLD7. A subsequent genome-wide association study conducted by a European consortium revealed strong associations with a noncoding SNP in PLA2R1 (rs4664308) and another in HLA-DQA1 (rs2187668), a member of HLA class II that includes isoforms that predispose carriers to autoimmunity.25 Although each of these two associated SNPs was significant alone, the odds ratio of MN was almost 80 in individuals who were homozygous for both HLA-DQA1 and PLA2R1 variants. Other genetic associations have been reported and may contribute to the severity or likelihood of MN progression.5 From 70% to 80% of all patients with MN present with the nephrotic syndrome.26,27 The remaining 20% to 30% present with subnephrotic asymptomatic proteinuria (<3.5 g/24 h). Proteinuria is nonselective. Microscopic hematuria is common (30% to 40%), but red blood cell (RBC) casts are rare and suggest a different glomerular pathologic process. In primary MN, serologic tests for anti-PLA2R are positive in 75% to 80% of cases,9,10,28 whereas serum complement levels are normal despite evidence of intraglomerular complement activation, and serologic markers (e.g., antinuclear antibodies, ANCA, rheumatoid factor) are normal or absent. At diagnosis, only 10% to 20% of MN patients have hypertension. Renal function is usually normal at presentation, with only a small fraction (<10%) presenting with renal impairment (Table 20-2). These presenting features can be modulated by age or preexisting hypertension; tubulointerstitial and vascular changes on biopsy may be related to these factors rather than to the severity of the MN.29 This is supported by recent evidence that age per se does not influence the rate of progression in MN, but does influence the GFR at presentation. Other complications related to the nephrotic syndrome include dyslipidemia, which probably contributes to the increased cardiovascular risk, and a high prevalence (10% to 40%) of thromboembolic events, including renal vein thrombosis. Recent large MN databases have indicated clinically relevant thromboembolic events occur in the range of 10% of nephrotic MN patients, most frequently in the first 1 to 2 years after presentation.30,31 The earliest pathologic feature of MN is the formation of subepithelial immune complexes of IgG and complement along the outer surface of the capillary wall in which glomeruli appear histologically normal and therefore may be mistaken for minimal change nephrotic syndrome, if only light microscopy is performed. MN begins with the formation of immune complexes at the interface of the podocyte and GBM, with subsequent changes in the podocyte, deposition of new extracellular matrix material between and around the immune deposits, thickening of the GBM (membranous change), and in some cases, focal glomerulosclerosis, tubular atrophy, and interstitial fibrosis.2,27 In the earliest stages of MN, the glomeruli and interstitium appear normal on light microscopy, and the diagnosis is made by immunohistology and electron microscopy (Fig. 20-4, A). The next stage of MN involves a homogeneous thickening of the capillary wall, seen with light microscopy in sections stained with hematoxylin and eosin or with periodic acid–Schiff (PAS) reagent (Fig. 20-4, B). On silver methenamine staining, early projections of the GBM between deposits may be detected in a characteristic spike-like configuration (see Fig. 20-4, C). Later, lucencies may develop in the GBM as immune deposits are resorbed, resulting in craters within the thickened GBM. Leukocyte infiltration is absent in glomeruli in MN, probably because chemotactic products of complement activation follow filtration forces into the urinary space rather than diffusing backward into the capillary lumen, and the intervening GBM prevents immune adherence mechanisms from being operative. As a result, the pathologic lesion of MN is characterized only by changes in podocytes and basement membrane, without associated glomerular hypercellularity. The podocyte response to this form of injury includes effacement of foot processes visible only by electron microscopy. In general, there are no visible mesangial or endothelial cell abnormalities. The presence of significant mesangial hypercellularity suggests immune deposit formation in the mesangium and is more consistent with a secondary MN, such as class V lupus nephritis (see Chapter 26). In some patients with heavy proteinuria and progressive disease, glomeruli exhibit reduced podocyte numbers and areas of focal sclerosis that are similar to the appearance of secondary focal segmental glomerulosclerosis (FSGS; see Chapter 18). These patients often have a more rapidly progressive course and a poor response to therapy. These sclerotic lesions may be a consequence of glomerular hypertrophy accompanied by an inability of terminally differentiated podocytes to proliferate,32 leading to areas of denuded GBM, attachment to Bowman capsule, and subsequent capillary collapse. As in all glomerular diseases, tubulointerstitial injury is common and correlates with both renal function and the level of proteinuria. Some studies suggest that the long-term outcome correlates in general with the severity of the tubulointerstitial damage (see Chapter 79). The pattern of granular glomerular capillary wall staining for IgG in MN is characteristic and easily recognizable by immunohistology (Fig. 20-5). Positive staining for IgG marks the finely granular subepithelial deposits, which are present on the outer surface of all capillary walls.2 The predominant IgG subclass in primary MN is IgG4.33–35 Positive staining for IgG1 or IgG3, IgA, or IgM or significant staining in the glomerular mesangium suggests lupus or other causes of secondary MN as an underlying mechanism.33–37 Kappa and lambda light-chain staining are typically equal but rare cases of MN due to monoclonal IgG, including anti-PLA2R have been reported.38 Complement C3 is also present in most cases of active disease and usually reflects staining for C3c, a breakdown product of C3b that is rapidly cleared. Consequently, positive C3 staining probably reflects active, ongoing immune deposit formation. When sought, staining for C5b-9 is generally present as well, consistent with the proposed pathogenetic role of C5b-9 in this disease.4 Strong C1q staining is not typically found in primary MN (<20% of cases)33,39,40 and is more common in lupus-associated MN.40 Positive staining for C4d in the absence of C1q is another feature of primary MN41,42 and may be consistent with activation of the mannose-binding lectin pathway of complement.43 Although not used in routine clinical practice, another feature that helps distinguish primary and secondary MN is the presence of PLA2R staining in the immune deposits in a pattern that co-localizes with IgG in anti-PLA2R-associated MN but not in secondary MN.9,12,44 Thus, several histopathologic features help distinguish primary and secondary forms of MN (Table 20-3). Table 20-3 Histopathologic features that help distinguish primary from secondary membranous nephropathy. Ig, Immunoglobulin; PLA2R, phospholipase A2 receptor. The presence of subepithelial electron-dense deposits by electron microscopy (EM) parallels IgG staining. In primary MN, immune deposit formation occurs in a subepithelial distribution; subendothelial deposits are not seen and mesangial deposits are rare (Table 20-3). These deposits in early stages of the disease process are homogeneous and may even be confluent in some areas, with overlying podocyte foot process effacement and little change in the underlying GBM (stage I). As the disease persists, basement membrane material is laid down between the deposits and corresponds to the spikes seen on light microscopy with use of a silver methenamine stain and are easily visible by EM (stage II; Fig. 20-6, A). Later, the spikes extend and the deposits may become surrounded by new basement membrane–like material (stage III; Fig. 20-6, B
Membranous Nephropathy
Definition
Etiology and Pathogenesis
Experimental Membranous Nephropathy
Human Membranous Nephropathy
Epidemiology and Genetics
Clinical and Serologic Manifestations
Pathology
Light Microscopy
Immunohistology
Distinguishing Histopathologic Features of Primary Versus Secondary Membranous Nephropathy
Primary
Secondary
Immunofluorescence Microscopy
IgG4 > IgG1, IgG3
IgG1, IgG3 > IgG4
IgA, IgM absent
IgA, IgM may be present.
Mesangial Ig staining absent
Mesangial Ig staining may be present.
C1q negative or weak
C1q positive
PLA2R positive and co-localizes with IgG
PLA2R negative
Electron Microscopy
Subepithelial deposits only ± mesangial deposits rarely
Subepithelial deposits ± mesangial and subendothelial deposits
Electron Microscopy
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Membranous Nephropathy
Chapter 20