Edward A. Ross, Kevin Damman
Management of Refractory Heart Failure
Nephrologists are being increasingly consulted regarding fluid management in patients with refractory heart failure. Renal expertise extends beyond diuretics and electrolyte homeostasis and now includes recent advances in acute and chronic fluid removal by extracorporeal dialysis machines or isolated ultrafiltration (UF) devices, as well as peritoneal modalities.
Definition and Scope of the Problem
Heart failure and renal dysfunction may coexist, but each disease can also cause or exacerbate the other. Poor cardiac output results in reduced renal perfusion and may result in kidney ischemia and progressive chronic kidney disease (CKD).1 Conversely, CKD may result in salt and water retention with subsequent venous congestion, hypertension, activation of the renin-angiotensin-aldosterone system (RAAS), and vascular calcification that cause cardiac dysfunction, accelerated atherosclerosis, and left ventricular hypertrophy and remodeling. These conditions subsequently may exacerbate heart failure, which results in a vicious cycle of reduced cardiac output and kidney dysfunction.
Of the 100,000 patients in the Acute Decompensated Heart Failure National Registry (ADHERE) 57% had CKD stages 3 or 4, 7% were at stage 5 (68% receiving dialysis), and only 9% had normal kidney function. In patients with heart failure who presented with CKD at the time of admission, there was greater use of diuretics and inotropes, less angiotensin-converting enzyme (ACE) inhibitor and angiotensin receptor blocker (ARB) administration, and a 5% higher in-hospital mortality rate.2
The cardiorenal syndrome has been classified3 into several categories, including cardiorenal (heart causing kidney disease), renocardiac (CKD leading to heart failure), and syndromes in which both are primary disorders or secondary to systemic conditions, although the significance of this classification for treatment strategies is unclear.
Pathogenesis
Figure 75-1 illustrates the pathogenesis of refractory heart failure.
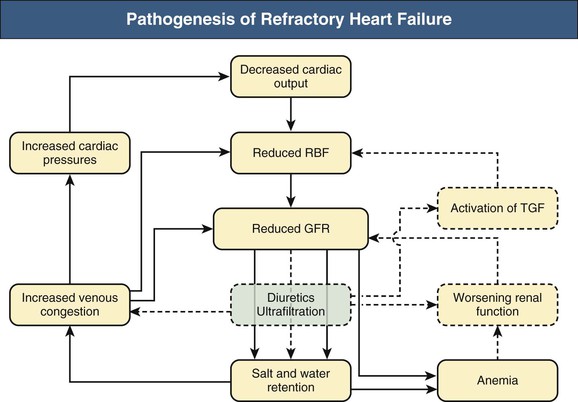
In the cardiorenal syndrome low cardiac output is the most important aspect of heart failure. Low cardiac output worsens renal perfusion and function, activating the RAAS and other systems, leading to salt and water retention with a subsequent and paradoxical further worsening of cardiac function. Central to this vicious cycle is tubuloglomerular feedback (TGF; described in Chapter 2), which is maladaptive in heart failure and in the case of some drugs that alter tubular sodium delivery. Secondary (hyper)aldosteronism (resulting in increased sodium retention), increased systemic vascular resistance (thereby putting more strain on the heart), and higher cardiac filling pressures may reduce cardiac output as described by the Frank-Starling mechanism, wherein further increase in preload reduces stroke volume. This further activates the sympathetic nervous system (SNS), which in turn worsens vasoconstriction, cardiac function, and renal perfusion. Excessive activation of both the RAAS and SNS pathways has long been considered the hallmark of worsened heart failure. However, other pathophysiologic mechanisms that could have important therapeutic implications have now been elucidated and are discussed in the following paragraphs.
Venous Congestion
There has been a growing appreciation that through multiple pathways right-sided congestion and renal venous hypertension cause kidney dysfunction.4 In this regard, heart failure would be analogous to such disorders as the abdominal compartment syndrome, portal hypertension, or renal venous thrombosis; however, some of these conditions differ in whether there is extravascular versus intravascular hydrostatic pressure transmitted to the kidneys. Venous hypertension is thought to trigger neuromyogenic changes that decrease renal perfusion, raise kidney interstitial pressure, narrow the arterial-to-venous pressure gradient, lower renal blood flow and glomerular filtration rate (GFR), trigger maladaptive autoregulatory responses, and exacerbate neurohumoral pathways already problematic in heart failure. Furthermore, there is also a direct effect of higher renal venous pressure that transmits to the renal parenchyma. This higher pressure directly opposes filtration in the Bowman capsule, causes collapsing of tubules, and may be the first initiating trigger for tubulointerstitial fibrosis.5 These mechanisms are consistent with a critical role of the adrenergic system in modulating venous tone and thus capacity. In that veins contain 70% of the circulatory volume, changes to venous capacitance (and pressure) can decompensate or ameliorate heart failure, without changes to total body salt, fluid, or weight.6 This variation in the venous reservoir volume reconciles how approximately a third of outpatients have heart failure decompensation with minimal change in weight, and how 16% of heart failure inpatients can symptomatically improve despite no weight loss. If validated, this pathway could potentially lead to pharmacologic treatments based on modulation of adrenergic tone. Changes in reservoir capacity would also confound interpretation of simultaneous changes in volume by extracorporeal UF. The importance of congestive renal failure is highlighted by studies in which kidney function in decompensated heart failure correlated better with right-sided pressures than the ejection fraction or cardiac index. In addition, elevated central venous pressures correlated with mortality.7 Better kidney function resulted when central venous pressures below 8 mm Hg were achieved.8
Adenosine Mediators
Adenosine receptors are involved with TGF signaling and maintain intrarenal vascular tone via a number of complex pathways; these include A1 receptors (A1Rs), which cause afferent arteriolar vasoconstriction, and A2 receptors, which induce efferent vessel dilation. Actions of adenosine at the macula densa and mesangium depend on angiotensin, renin, nitric oxide, and prostaglandin levels. A1R antagonists could thereby not only restore diuretic sensitivity but also allow appropriate aquaresis in severe heart failure.
Inflammatory Cytokines
Chronic as well as acute exacerbations of heart failure are associated with widespread inflammation and high levels of proinflammatory cytokines, which may further boost cardiac, renal, and other tissue dysfunction.
Anemia
Anemia may be caused by chronic disease or coexistent CKD, or it may be the consequence of hemodilution by venous congestion. The term cardiorenal anemia syndrome emphasizes this pharmacologically remediable aspect of heart failure.
Diuretic Tolerance and Adverse Effects
Many patients with heart failure develop tolerance to chronic diuretic therapy, fail to have appropriate natriuresis despite escalating doses, and have worsening of neurohumoral mediator levels—that is, diuretic resistance.9 Thus some of the cardiac benefits from ACE inhibitors might be from blocking diuretic-induced RAAS activation. Consistent with this paradigm and data from the ADHERE registry, morbidity and mortality in advanced heart failure increased up to fourfold with higher doses of diuretics, despite the fact that there were no clinical or echocardiographic parameters to suggest that the high-dose group had any worse cardiac function.10 However, when differences in baseline characteristics were controlled for, outcomes with respect to diuretic therapy were similar in a large French registry of patients with acute heart failure.11 In a multicenter analysis, patients on high-dose diuretics had worsening of GFR compared with the control group on alternative medications, despite equivalent fluid losses.12 Finally, in the Diuretic Optimization Strategies Evaluation (DOSE) trial (see later), there was no difference in outcome in high- versus low-dose diuretic therapy, despite a higher incidence of (transiently) worsening GFR in the high-dose diuretic group.13
Treatment
General Approach and Limitations
A pragmatic approach is to first address potentially treatable problems such as valvular disease, conduction disorders and arrhythmias, pericardial effusion, or coronary ischemia (e.g., by angioplasty and stenting) (Box 75-1). The clinician can then approximate the volume of excess fluid and can craft a daily therapeutic goal concurrent with salt restriction. Medication-naive patients can then be cautiously treated as described in the following sections, with serial monitoring because hypotension or renal dysfunction commonly limits the use of pharmaceuticals. Determining to what degree a particular patient may be able to tolerate drug-induced hypotension (e.g., from afterload-reducing agents) is clinically challenging. If there is marked sensitivity of the blood pressure or GFR to low doses of RAAS blockade, bilateral renal artery stenosis should be considered (see Chapter 39).
Pharmacologic Therapeutic Strategies
The pharmacologic management of heart failure is summarized in Box 75-1 and includes traditional approaches for arrhythmias (e.g., for atrial fibrillation) and afterload reduction, SNS blockade, and mineralocorticoid receptor antagonism. Although diuretics remain the front-line treatment for heart failure patients, adequate salt and water clearance in acute heart failure is often difficult; registry data indicated that of patients admitted with acute heart failure, approximately 16% were discharged at a higher body weight.14
Diuretics
Despite the concerns described previously, most patients initially respond to escalating doses of diuretics. Loop diuretics need to be administered at frequent intervals at a high enough dose to achieve adequate drug levels within the glomerular filtrate. Coadministering a long-acting thiazide diuretic may help maintain natriuresis. In severe heart failure, continuous low-dose infusion of a loop diuretic may be required. In the DOSE study,13 high versus low doses and continuous versus bolus infusion of loop diuretics were tested in acute heart failure. None of the dosage regimens was associated with superior outcome. Although higher dosages of diuretics resulted in more frequent transient decreases in GFR, this did not affect the long-term outcomes. Based on the recently completed Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial, in which there was worsened renal function in decompensated heart failure, efficacious and safe algorithms can be crafted for stepped pharmacologic care wherein inotropes are added to diuretics.15
Renin-Angiotensin-Aldosterone System Antagonists
Although low GFR is a marker for increased mortality with heart failure, a further fall in GFR after initiation of RAAS blockade does not necessarily portend a worsened outcome.16
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


