Sickle Cell Disease
Vimal K. Derebail
Abhijit V. Kshirsagar
Although reports suggestive of the disease that would come to be known as sickle cell anemia were published in the 1800s,1,2 Dr. James B. Herrick is credited with the first modern description in 1910. Herrick and his intern Ernest E. Irons described the case of Walter Clement Noel, a male dental student from Grenada (West Indies), who originally sought care for a cough and fever and whose blood film revealed what Irons described as “peculiar elongated and sickle-shaped red blood corpuscles.”3,4 In this first report, Herrick went on to describe isosthenuria, now recognized as one of the most common manifestations of sickle nephropathy.
Current understanding of sickle cell disease reveals it to be a genetic disorder that follows a pattern of Mendelian autosomal co-dominant inheritance5,6 and affects numerous organ systems. Molecular studies have demonstrated the defect to be with the hemoglobin molecule,7 specifically, the result of a substitution of the amino acid valine for glutamic acid at position 6 of the β chain.8,9,10 A high prevalence of sickle cell disease and heterozygous sickle hemoglobin exists in areas with a high burden of malaria, including sub-Saharan Africa, the Arabian peninsula, and parts of the Indian subcontinent. The presence of hemoglobin S (HbS) has long been postulated to confer a protective benefit to infection by Plasmodium falciparum, making carriers less susceptible to malarial morbidity and mortality. This characteristic offers possible explanation for the global distribution and persistence of the mutation.
This chapter focuses on renal abnormalities caused by the presence of sickle hemoglobin. Since Herrick’s initial description of isosthenuria, a greater understanding of the clinical effects of sickle hemoglobin on kidney morbidity has been gained. The chapter also includes a discussion of the rare but lethal cancers seen among individuals with sickle cell disease. In addition, the chapter describes the potential role of heterozygote inheritance of hemoglobin S in renal disease.
GENETICS
Several decades following Herrick’s description, Linus Pauling noted differing electrophoretic properties of hemoglobin taken from individuals with sickle cell anemia in 1949.7 With this discovery, it became known as the first “molecular disease.” Shortly thereafter, Vernon Ingram and James Hunt identified the exact nature of the molecular alteration—the substitution of valine for glutamic acid at the sixth residue of the β chain of hemoglobin.9,10,11
The genetic mutation responsible for the single amino acid substitution occurs on the short arm of chromosome 11, and asserts as an autosomal recessive pattern of inheritance. The mutation itself results from a single nucleotide substitution from a GAG to GTG codon mutation. Because of incomplete recessive inheritance, those with a single mutation (i.e., carriers with sickle cell trait), produce a measurable quantity of hemoglobin S.
The hemoglobin S mutation seems to have arisen in at least two separate occurrences, one in Africa and the other in Asia. In fact, there are four African specific β-globin gene haplotypes—the Benin, Cameroon, Senegal, and Central African Republic (CAR) haplotypes—and one specific to Asia-the Arab-Indian haplotype.12,13 These several variations in the β-globin gene mutation do seem to confer variance in the observed phenotype. Those individuals with the Senegal haplotype seem to have the least severe disease course, whereas those with the CAR haplotype have the worst disease manifestations.13 Worldwide, sickle cell disease is estimated to affect over 275,000 live births annually, and over 300 million individuals are carriers of the gene for sickle hemoglobin. In North America and Europe, 2,600 and 1,300 individuals, respectively, are born yearly with these disorders, whereas in southeast Asian regions and Africa these numbers reach 26,000 and 230,000, respectively.14 These disease frequencies closely parallel the distribution of malaria, prior to the interventions to control its spread.12
PATHOPHYSIOLOGY
Normal adult hemoglobin (HbA) is a 68 kilodalton molecule composed of two pairs of α and β polypeptide chains folded around an iron-containing heme ring. Fetal hemoglobin (HbF), consisting of α and γ chains, predominates during in utero development. The β chain synthesis overtakes γ chain
production shortly after birth and the hemoglobin tetramer takes on adult form.
production shortly after birth and the hemoglobin tetramer takes on adult form.
The result of the point mutation in the β chain is a modest change in the three-dimensional spatial configuration but a profound change in the solubility of hemoglobin. Glutamic acid is a charged amino acid and as a polar molecule, highly soluble in water. Valine is uncharged, making it poorly soluble in water. The entire sickle hemoglobin molecule takes on the property of the valine amino acid, becoming nonpolar and poorly soluble in water. The loss of charge, incidentally, explains the altered migration of sickle cell hemoglobin during electrophoresis.
The clinical sequelae of sickle cell disease, including kidney manifestations, arise from the properties of the hemoglobin resulting from the glutamine to valine amino acid substitution of the β subunit. The key steps that lead to organ injury include (1) polymerization of the sickle hemoglobin; (2) adhesion of affected red blood cells; and (3) ischemic/ oxidative injury to the vasculature. The degree of injury to any organ system, including the kidney, depends on a number of factors that promote or inhibit these key steps.
Left unimpeded, the uncharged, poorly soluble valine residues allow the hemoglobin molecules to adhere to one another. Polymerization of the hemoglobin S results from the continued aggregation the insoluble molecules. Classically, seven pairs of sickle hemoglobin chains aggregate to form large, insoluble polymers.15 The rate of sickle hemoglobin polymerization has been proposed to determine the ultimate morphology of the red blood cell16; slow polymerization results in long sickle hemoglobin fibers with classic sickle shape whereas rapid polymerization leads to shorter, more granular fibers. Once polymerized, sickle hemoglobin forms a highly viscous, semisolid “gel”16 with a solubility much less than hemoglobin A and forms elongated structures that distort the red cells into their characteristic sickle and other abnormal shapes (Fig. 62.1).
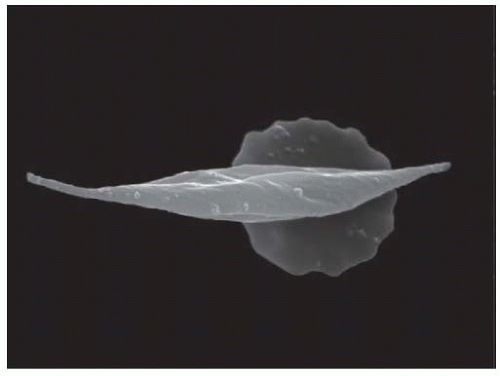 FIGURE 62.1 Normal red blood cell (background) and red blood cell affected by sickle-cell anaemia (foreground). Wellcome Library, London, “Sickle Cell Anemia” under Creative Commons by-nc-nd 2.0 UK: England & Wales, with permission from the Wellcome Trust. (See Color Plate.) |
The polymerization of sickle hemoglobin leads to mechanical deformation of the red blood cell. Acquired red blood cell membrane abnormalities result, including a loss of phospholipids, asymmetry, increased fragility, and abnormal integrin-protein interactions.17 This injury, in addition to the retention of adhesion molecules, promotes sickle red blood cell adhesion to vascular endothelium.18,19 Furthermore, the presence of hemoglobin S accentuates red cell endothelial adhesion,20,21 potentially producing stasis and occlusion in slow-flowing renal vessels under even mild physiologic stress. Clinical disease severity has been correlated with red blood cell adhesivity.22
Oxidative injury related to free heme exposure is a notable feature of the pathology of renal injury. Heme oxygenase (HO), the rate-limiting enzyme in heme degradation, serves as a measure of oxidative stress. Studies in transgenic murine models and in humans have demonstrated renal induction of heme-oxygenase-1 (HO-1), the isozyme induced by oxidative stress (Fig. 62.2). Inhibition of glutathione synthesis in the same transgenic murine model to produce acute oxidative stress precipitated acute vaso-occlusive episodes in the kidney, further demonstrating the role of oxidative stress to renal pathology.23
Physiologic, metabolic, and genetic conditions promote and mitigate the polymerization process and may help to explain the variability of clinical disease. In vitro studies have demonstrated that decreased oxygen concentration, increased hydrogen ion concentration, rapid deoxygenation, low temperature, advanced red blood cell age, high osmolality, and high intracellular HbS concentration promote the formation of the sickle polymers.24,25,26,27,28,29
Two well-established genetic modifiers of phenotypic severity are fetal hemoglobin concentrations and coinheritance of α-thalassemia. Fetal hemoglobin concentration may range between 1% and 30% and is determined by at least three different genetic loci.12 With higher levels of fetal hemoglobin, the concentration of hemoglobin S in the erythrocyte is reduced. Nearly 30% to 50% of patients with sickle cell disease have coexistent α-thalassemia trait, which leads to an overall reduction in the concentration of total erythrocyte hemoglobin and reduced hemoglobin S polymerization.12 Both of these modifying genetic factors have been shown to modulate the severity of clinical manifestations.12
Normal physiologic functions of the kidney include excretion of acid and solute. Gradients of acid and solute occur as a result of counter-current transport, and render the medulla with especially high concentrations of both. Furthermore, the renal medulla has a very low partial pressure of oxygen (10-20 mm Hg),30 sufficient to induce sickling.31 In aggregate, the conditions of the renal medulla—relative hypoxia, hyperosmolarity, acidity, and sluggish blood flow (Fig. 62.3)—represent an environment primed for sickling of red blood cells containing hemoglobin S. Angiographic studies have demonstrated significant renal vascular disruption and vessel dropout in individuals with HbSS and HbAS compared to individuals with normal hemoglobin
(HbAA)—likely the result of repeated episodes of red blood cell sickling with resultant ischemia (Fig. 62.4).32 Experimental mouse models of sickle cell anemia have demonstrated an exquisite sensitivity to ischemic injury.33,34 Chronic hypoxia may mediate the development of focal segmental glomerular scarring and subsequent proteinuria notable in sickle cell disease.
(HbAA)—likely the result of repeated episodes of red blood cell sickling with resultant ischemia (Fig. 62.4).32 Experimental mouse models of sickle cell anemia have demonstrated an exquisite sensitivity to ischemic injury.33,34 Chronic hypoxia may mediate the development of focal segmental glomerular scarring and subsequent proteinuria notable in sickle cell disease.
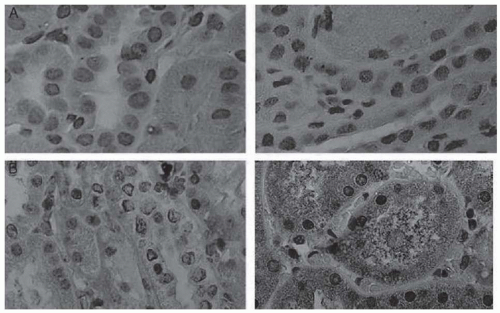 FIGURE 62.2 Immunoperoxidase staining for HO-1 in normal human kidney (A) and in the kidney of a patient with sickle cell disease (B). In A and B, immunoperoxidase studies were undertaken in the absence (left) and presence (right) of HO-1 antibody. B: Positive staining with the HO-1 antibody in renal proximal tubules in the kidney of the patient with sickle cell disease (right). Original magnification, ×400. (From Nath KA, Grande JP, Haggard JJ, et al. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol. 2001;158:893-903, with permission from Elsevier at the courtesy of the editors.) |
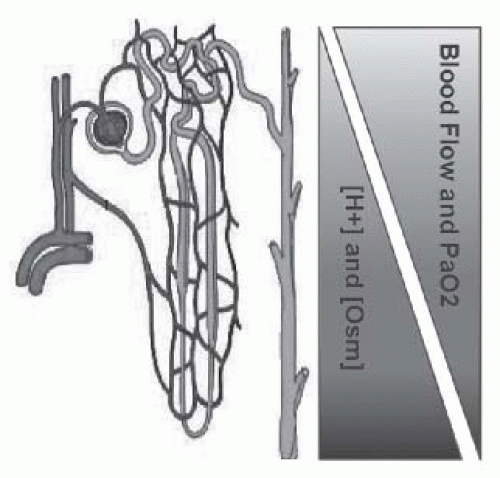 FIGURE 62.3 The renal medulla produces an environment in which red blood cells containing hemoglobin S are more likely to undergo sickling. As red blood cells traverse the renal vasculature into the medulla, they enter an area of increasing acidity and osmolarity and decreasing oxygen content and blood flow. |
Recent investigations into the pathophysiology have also demonstrated a role for endothelial dysfunction. Patients with sickle cell disease demonstrate high levels of soluble fms-lke tyrosine kinase-1 (sFlt-1), which is a member of the vascular endothelial growth factor (VEGF) receptor family. High levels of sFlt-1 are felt to promote endothelial dysfunction by binding to the receptor-binding domains of circulating VEGF and preventing the normal interaction with endothelial cell receptors. A recent cohort study of 73 patients with sickle cell disease demonstrated statistically higher values of sFLT-1 among those patients with albuminuria, suggesting it may induce glomerular endothelial dysfunction.35
Additional investigations into the pathophysiology of chronic kidney disease (CKD) have gone beyond previous understanding of genetic modifiers as mentioned previously and have focused on the role of coinherited gene polymorphisms in the development of kidney disease.36 Specifically, investigators have focused on the transforming growth factor beta and bone morphogenetic protein (TGF-β/BMP) pathway, implicated in the development of diabetic nephropathy and also associated with other organ involvement in sickle cell anemia.37,38,39,40,41,42 In a longitudinal analysis of over 1,000 individuals with sickle cell anemia, the authors found that
four distinct single nucleotide polymorphisms (SNPs) in the BMPR1B, a receptor gene, were significantly associated with GFR. Utilization of genomewide association studies (GWAS) has identified further SNPs that may be important to explaining the phenotypic heterogeneity of sickle cell disease.43 Additional analyses are needed to confirm promising exploratory studies that demonstrate modulation of renal morbidity among individuals with sickle cell anemia.
four distinct single nucleotide polymorphisms (SNPs) in the BMPR1B, a receptor gene, were significantly associated with GFR. Utilization of genomewide association studies (GWAS) has identified further SNPs that may be important to explaining the phenotypic heterogeneity of sickle cell disease.43 Additional analyses are needed to confirm promising exploratory studies that demonstrate modulation of renal morbidity among individuals with sickle cell anemia.
 FIGURE 62.4 Injection microradioangiographs of kidneys from a subject without hemoglobinopathy (A), a patient with sickle cell disease (B), and a patient with sickle cell-hemoglobin C disease (C). In the normal kidney (A), vasa recta are visible radiating into the renal papilla; in sickle cell anemia (B), vasa recta are virtually absent. Those vessels that are present are abnormal, are dilated, form spirals, and end bluntly, and many appear to be obliterated. The patient with Hb-SC (C) shows changes intermediately between the Hb-SS patients and the normal subjects. (Reprinted from Statius van Eps LW, et al. Nature of concentrating defect in sickle cell nephropathy, microradioangiographic studies. Lancet. 1970;1:450, with permission from Elsevier at the courtesy of the editors.) |
RENAL MANIFESTATIONS
Structural Changes
Renal Size
The kidneys of patients with sickle cell disease typically vary in size with age. Infants and young children have kidneys of near-normal size.44 The kidneys increase in length and weight in older children and young adults—on average the kidneys growing by 5 to 8 mm per year by ultrasound imaging in patients with Hb-SC and Hb-SS disease as compared with age-matched controls.45 After age 40 years, the size decreases.46
Other structural changes are commonly seen on radiographic imaging. These findings increase with advancing age and likely reflect the cumulative effect of repeated episodes of “sickling.” Cortical scarring is seen on intravenous pyelography among 8% of individuals aged 16 to 25 years, and rising to 45% among those over 35 years.47 Calyceal abnormalities such as blunting, cysts, and clubbing are seen among 28% of youths and among 57% of individuals over 35 years.47 Papillary necrosis among adults is seen at high frequency, ranging from 15% to 65%.48,49,50 B-mode ultrasound imaging can reveal increased echogenicity of the kidneys in up to 25% of individuals with sickle cell anemia.51,52
Kidneys removed due to severe hematuria may demonstrate submucosal hemorrhages in the pelvis, medulla, and cortex.53 Radiographic changes suggesting papillary necrosis, medullary cavitations, ring shadows, and calcifications in the pyramids have also been observed.54,55 Occasionally, minimal papillary necrosis is apparent only on microscopic examination. Renal vein thrombosis is also an occasional finding in Hb-SS disease. Finally, the macroscopic appearance of kidneys of affected individuals may show irregularity and granularity of the surface.
Vasculature
Microradioangiographic studies (Fig. 62.4) have been performed on kidneys removed at autopsy from patients with normal hemoglobin, patients with Hb-SS disease, and patients with Hb-AS and Hb-SC disease.32 A significantly reduced number of vasa recta are seen in kidneys from Hb-SS patients. The vessels that remain are abnormal as they are dilated, show spiral formation, and end bluntly. Patients with Hb-AS and Hb-SC disease show changes intermediate between those of the Hb-SS patients and normal subjects. In patients with Hb-AS, sparse bundles of vasa recta are surrounded by a chaotic pattern of dilated capillaries, with loss of the original bundle architecture. The changes presumptively result from occlusion of vasa recta and represent the structural basis for the development of functional changes. The loss of the highly specialized structure of parallel running loops of Henle and vasa recta disrupts the normal countercurrent multiplication and exchange.
Histology
Glomerular
Enlargement of the glomeruli is a common finding in patients with sickle cell disease. The glomeruli may be visible by the naked eye and have been described as red “pinheads.”27,56 Individuals with homozygous sickle hemoglobin have both an increase in glomerular diameter and size compared to normal controls.57,58 Glomerular enlargement and congestion are more common in children beyond the age of 2 years and are most marked in juxtamedullary glomeruli.57,59 When the size of juxtamedullary glomeruli is systemically measured, there is a distinct difference between those of children with sickle cell disease and normal children. In older patients, this glomerular enlargement and congestion may lead to progressive ischemia and fibrosis with obliteration of glomeruli.46,60
Histology of the enlarged glomeruli is notable for the marked hypercellularity and lobulation of the glomerular tuft. Other changes include replication of the basement membrane and mesangial proliferation.57,61,62 Aggregates of sickled red blood cells may pack and distend the glomerular capillaries and the afferent and efferent arterioles. Electron microscopy of glomeruli may reveal foot process effacement and local thickening of the basement membrane,61 although an exact prevalence is difficult to ascertain.
Clinical syndromes resulting from glomerular pathology, including glomerulonephritis, have been described among individuals with sickle cell disease. These conditions are discussed later in the chapter.
Medullary
The major components of the medulla are the vasa recta and renal tubules. As noted earlier, physiologic factors favoring sickling are routinely found in the medulla and, as such, medullary lesions are among the earliest and most prominent renal abnormalities. Initial changes consist of edema, focal scarring, and interstitial fibrosis. Progressive scarring leads to tubular atrophy and infiltration of mononuclear cells. Iron deposition has been observed in proximal tubules and pigmented casts may be seen.55,57,61 Defective iron metabolism, as suggested by decreased renal cortical spin echo with magnetic resonance imaging (MRI), may contribute to the nephrotic syndrome.63 All these changes could be the result of the observed obliteration or attenuation of the medullary circulation.32
Functional Changes
Renal Hemodynamics and Function
Renal hemodynamics are thought to be either normal or supernormal in homozygous (Hb-SS) patients younger than 30 years of age.64,65,66 In Hb-SS infants, increased values have been observed for both GFR and effective renal blood flow (ERBF), as well as for the tubular transport maximum of para-aminohippurate (TmPAH). A recent study of a cohort of infants with sickle cell disease has demonstrated similar findings of increased GFR.67,68 ERBF has been reported to be normal or elevated, although less elevated than effective renal plasma flow (ERPF) because of the typically very low hematocrit, while the extraction ratio of para-aminohippurate (EPAH) was decreased. Filtration fraction (GFR/ERPF) has been found to be decreased (mean 14% to 18%; normal 19% to 22%).64,65,69 Selective damage of the juxtamedullary glomeruli might result in a lower filtration fraction because these nephrons have the highest filtration fractions.70,71 Microradioangiographic studies lend support to this suggestion.32
In sickle cell trait, Hb-SC disease, Hb-CC disease, hemoglobin C trait, and Hb-Sβ+ thalassemia, GFR, and ERPF have been reported to be within the range of normal.72 As compared to patients with Hb-SS, studies in individuals with sickle cell trait do not appear to demonstrate significant age-related increases in GFR or microalbuminuria.73
Several mechanisms have been postulated for the increase in renal hemodynamics in patients with sickle cell disease. With severe anemia, peripheral vascular resistance decreases and cardiac output increases, possibly causing blood to be preferentially shunted to the renal circulation. However, in studies with short follow-up, correction of the anemia does not alter the GFR or ERBF.64,74 Multiple transfusions with Hb-AA blood to patients with Hb-SS result in significant, although temporary, increases in hemoglobin concentration, with gradual and almost complete replacement of Hb-S by Hb-A. Because this procedure does not reduce the supernormal GFR and ERPF,64 the cause of the supernormal renal clearances in sickle cell nephropathy cannot be explained by the anemia per se or by the presence of the abnormal hemoglobin. Altered nitric oxide production and renin secretion as seen in transgenic sickle cell mouse models may play a role in hyperfiltration.75,76 (See section entitled “Pathophysiology.”) The ischemic damage to the medulla could also be a stimulus for increased prostaglandin synthesis that may drive hyperfiltration.66,69,77 Administration of indomethacin, an inhibitor of prostaglandin synthesis, to individuals with sickle cell anemia results in a significant fall in GFR, ERPF, creatinine clearance, and urea clearance supporting this contention.69
A number of possible mechanisms may be responsible for the decline in renal hemodynamics with aging, sometimes ending in renal failure with shrunken end-stage kidneys at necropsy. Although advancing age has an effect on renal hemodynamics and overall kidney function in all individuals, these effects may be heightened in individuals with sickle cell anemia because of the disruption of the medullary vasa recta. Support for the idea of a reduction in medullary blood flow in sickle cell disease can be found in the microradioangiographic examination of Hb-SS kidneys (Fig. 62.4).32 In these studies of kidneys obtained at autopsy, perfusion of the vasa recta by contrast medium is virtually absent, suggesting an almost complete absence of vasa recta in Hb-SS. Supernormal hemodynamics and hyperfiltration have been proposed as causative mechanisms of glomerulosclerosis,78 which may lead to the chronic renal failure seen in sickle cell disease. Increased apoptosis, stimulated by ischemic and/ or oxidative injury, may also contribute to progressive renal dysfunction.79,80,81 Over a number of years, continued loss of medullary circulation, interstitial fibrosis, and possibly superimposed pyelonephritis may lead to a progressive decline in GFR and advanced renal insufficiency in the patient with Hb-SS disease. Thus, there is a progressive decline in ERBF, ERPF, and GFR,65,82 although there are rare individuals with apparently normal kidney function.
Clinically, the progressive loss of GFR is used to stage CKD. Stages 1 and 2 capture a GFR range above 60 mL/ min/1.73 m2 with proteinuria whereas stages 3 to 5 include a GFR range less than 60 mL/min/1.73 m2 and stage 6 denotes those receiving renal replacement therapy or dialysis. Although advanced renal failure—that which requires renal replacement therapy, or that which is ascribed as a cause of death—is well documented,83,84,85,86 estimates of earlier stages of renal failure vary widely87,88 and should be interpreted with caution. Death from nonrenal causes (infection, pulmonary disease, etc.) may be a competing risk, limiting detection of CKD or end-stage renal disease (ESRD). Determining CKD prevalence is further complicated by the fact that serum creatinine, derived from muscle tissue and routinely used to estimate GFR in a variety of formulas,89,90,91 may actually overestimate GFR determined by gold standard radionucleotide excretion studies in individuals with sickle cell anemia.92,93
Generation of Negative Solute-Free Water
The capacity to generate negative solute-free water (TcH2O) has been studied in patients with sickle cell disease using different protocols of mannitol loading with conflicting results. Although Whitten and Younes found normal TcH2O in Hb-SS children,94 Levitt et al. describe two patients with a TcH2O of 3.2 mL/min/100 mL glomerular filtrate.95 Hatch et al. similarly noted a mean TcH2O of 4.2 ± 0.9 SD mL/min/100 mL glomerular filtrate in 11 Hb-SS patients compared to a mean TcH2O of 5.7 ± 1.2 SD mL/min/100 mL glomerular filtrate in 7 control subjects.96 These results suggest that TcH2O after mannitol loading is impaired in sickle cell anemia.
Studies following the response to saline infusion96,97 demonstrate impaired TcH2O in Hb-SS patients. From these studies, sickle cell anemia patients were concluded to have a defect in the water impermeable medullary loops of Henle that transports solute. The normal solute-free water clearance under standard conditions argues against such
impairment in sodium chloride reabsorption from the ascending limb of the loop of Henle. However, in comparison to normal solute-free water clearance, a normal TcH2O depends on adequate function of the portion of the loop of Henle that is localized in the inner medulla. Sickle cell anemia patients are able to increase urinary osmolality generally to 450 mOsm per kg H2O or higher, to the level that can be generated in the outer medulla. As shown in Figure 62.4, the capillary plexus surrounding short loops of Henle in the outer zone does not necessarily penetrate into the inner medullary zone.
impairment in sodium chloride reabsorption from the ascending limb of the loop of Henle. However, in comparison to normal solute-free water clearance, a normal TcH2O depends on adequate function of the portion of the loop of Henle that is localized in the inner medulla. Sickle cell anemia patients are able to increase urinary osmolality generally to 450 mOsm per kg H2O or higher, to the level that can be generated in the outer medulla. As shown in Figure 62.4, the capillary plexus surrounding short loops of Henle in the outer zone does not necessarily penetrate into the inner medullary zone.
Urinary Diluting Capacity
Patients with sickle cell anemia are capable of diluting their urine normally.74,96,98 Under conditions of water diuresis, the fall in urinary osmolality and percentage of filtered water excreted (CH2O/GFR) has been found to be the same in Hb-SS patients when compared to99,100 controls. Therefore, the capacity to reabsorb solute in the thick portion of the medullary ascending limb of the loop of Henle apparently is intact in sickle cell anemia.
This combination of a defect in renal concentrating capacity with a normal diluting capacity is quite characteristic for sickle cell anemia. However, unlike control subjects, the normal diluting capacity of sickle cell patients may be particularly dependent on prostaglandins. In one series, administration of indomethacin, an inhibitor of prostaglandins, led to a greater fall in CH2O/GFR in sickle cell subjects and a rise in urinary osmolality from 42 to 125 mOsm per kg H2O (Fig. 62.5).99,100 These results suggest that impairment of renal prostaglandins may hamper the normal diluting capacity in sickle cell anemia.
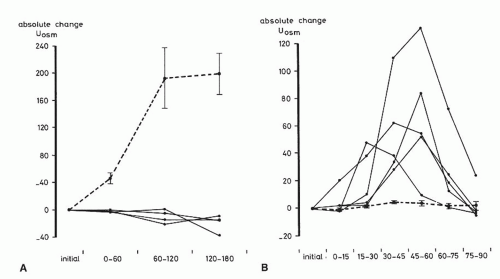 FIGURE 62.5 The effect of indomethacin (75 mg as a suppository) in the water-depleted state (A) and the effect of indomethacin (0.25 mg/kg body weight intravenously) in the water-loaded state (B). The broken lines represent the mean ± standard error of the mean in control subjects, and the continuous lines represent the individual data in patients with sickle cell anemia. (Reproduced with permission from De Jong PE, et al. The influence of indomethacin on renal concentrating and diluting capacity in sickle cell nephropathy. Clin Sci. 1982;63:53, © the Biochemical Society.) |
Urinary Acidification
Although systemic acidosis is generally not a feature of sickle cell disease in the absence of advanced renal failure, patients with Hb-SS or Hb-SC may demonstrate an incomplete form of renal tubular acidosis.101,102,103,104 In response to a shortduration acid load, between 29% and 100% of patients with Hb-SS101,102,103,104 are unable to decrease urine pH below 5.3, whereas normal subjects can achieve a urinary pH of 5.0 or lower. Titratable acid and total hydrogen ion excretion are lower in patients with Hb-SS and Hb-SC, but ammonia excretion is appropriate for the coexisting urine pH in most cases. The increased ammonia excretion induced by acid loading is also impaired by indomethacin,101 suggesting the assumed enhanced prostaglandin synthesis in sickle cell disease105 may also be important to maintaining normal ammoniagenesis in sickle cell disease. When a maximal acidifying stimulus is employed, such as infusion of sodium sulfate, patients with Hb-SS may lower urine pH and increase net acid excretion to the same degree as normal subjects. Thus, the distal tubule of Hb-SS patients apparently requires a greaterthan-normal stimulus to generate a normal urine-to-blood hydrogen ion gradient. In contrast, renal acidification has been found to be normal in patients with sickle cell trait.106
The acidification defect has been classified as distal rather than proximal103 and is characterized by failure to achieve a normal minimal urinary pH with acid loading but is not associated with bicarbonate wasting. Because patients studied not were acidemic or hyperchloremic before acid loading and no generalized proximal tubular reabsorptive defect was observed, the acidification defect was consistent with an incomplete distal renal tubular acidosis.106 Speculatively, alterations in microcirculation of the renal papillae may impair maintenance of the normally steep hydrogen ion gradients in the collecting ducts. This very subtle defect in renal acidification generally does not cause a systemic metabolic acidosis, which could increase sickling. Several studies globally102,104,107,108 have been unable to demonstrate evidence of metabolic acidosis in the absence of a sickle cell crisis, but do exhibit changes consistent with a mild chronic respiratory alkalosis.
Potassium Metabolism
In addition to the defect in hydrogen ion excretion, potassium excretion may also be impaired in sickle cell patients.109,110,111 Following potassium chloride, sodium sulfate, or furosemide administration, potassium excretion was found to be subnormal in patients with sickle cell disease. Hypoaldosteronism does not appear to explain these findings as both plasma renin activity and plasma aldosterone concentration were normal, both during normovolemia and after volume contraction. Additionally, despite impaired renal potassium excretion, hyperkalemia did not develop in these patients during acute potassium chloride loading. Although administration of angiotensin-converting enzyme (ACE) inhibitors and other agents may elevate plasma potassium concentration,112
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis




Related posts:
Computed Tomography and Magnetic Resonance Imaging
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Pathophysiology of Nephrotoxic Cell Injury
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
