Stephen C. Textor, Barbara A. Greco
Renovascular Hypertension and Ischemic Nephropathy
Few areas in nephrology generate more controversy than management of renovascular disease. Advances in medical therapy, management of atherosclerotic comorbidity, diagnostic imaging, and endovascular intervention have combined to reshape the tools available and the range of clinicians involved with managing these disorders. Restoring blood flow to a critically ischemic kidney offers the potential to improve blood pressure and recover kidney function. However, several recent prospective, randomized controlled trials have failed to identify major additional benefits from revascularizing the kidney when added to optimal medical therapy. Although these trial results have diminished enthusiasm for renal artery revascularization for many patients, nephrologists will continue to encounter patients who can benefit from endovascular or surgical intervention for advanced renovascular disease as well as intensive risk factor management. As with many vascular diseases, the issue is most often one of timing, rather than simply choosing one therapy over another indefinitely. This chapter provides an overview of the pathophysiology of renovascular hypertension and clinical management of renal artery stenosis.
Definition and Etiology
Renovascular disease with reduced perfusion of the kidney can produce a range of clinical syndromes, most frequently a rise in arterial pressure, designated renovascular hypertension (RVH), with or without associated ischemic and hypertensive renal injury. RVH is usually caused by renal artery (RA) stenosis and is a common secondary form of hypertension. Recognition that reduction of renal perfusion pressure activates a series of hormonal and neuronal responses that raise systemic arterial pressure remains one of the seminal observations in blood pressure (BP) regulation. Not surprisingly, high BP in patients with RA stenosis frequently leads to the assumption that the stenosis is the cause of the hypertension. Often, however, patients have preexisting hypertension unrelated to renovascular disease. Final proof that a patient has RVH rests with the demonstration that the hypertension is improved or eliminated by RA endovascular or surgical revascularization or by removal of the kidney distal to the stenosis.
Renovascular hypertension is defined as a syndrome of elevated BP (systolic and/or diastolic) produced by any condition that interferes with arterial circulation to the kidneys. The majority of patients with RVH have significant main RA stenosis with reduced renal perfusion pressure. Most conditions cause reduced perfusion to one kidney, while a second “contralateral” kidney is exposed to elevated systemic pressures, called “two-kidney” hypertension by analogy to experimental models of “2-kidney–1-clip hypertension” discussed later. When both kidneys are affected, as may occur with atheroembolic disease or a RA stenosis in a solitary functioning kidney without a normal “contralateral” kidney, the designation “one-kidney” RVH is given.
Of the conditions that may produce the syndrome of RVH, main vessel RA stenosis is by far the most common. The two major causes of main renal artery disease are fibromuscular dysplasia (FMD) and atherosclerotic renal vascular disease (ASRVD). Box 39-1 lists other conditions that can cause RVH by impairing renal blood supply. Many of these are rare, but all lead to reduced renal perfusion pressure.
The term ischemic nephropathy or ischemic renal disease (IRD) refers to the reduced glomerular filtration rate (GFR) associated with reduced renal blood flow beyond the level of renal autoregulatory compensation.1 Critical RA stenosis can lead to renal atrophy and progressive renal impairment. As with RVH, establishing the causal relationship between proximal RA stenosis and the development and progression of chronic kidney disease is often difficult. Collateral renal blood supply can preserve renal viability in the face of proximal atherosclerotic renovascular occlusive disease, and small vessel and parenchymal disease often coexist with main RA atherosclerotic disease. Renal revascularization of ischemic kidneys in some cases can succeed in recovering renal function, but there are few predictors to guide therapeutic choices in these patients, who are often at high risk for complications of interventions.
Pathophysiology of Renovascular Hypertension
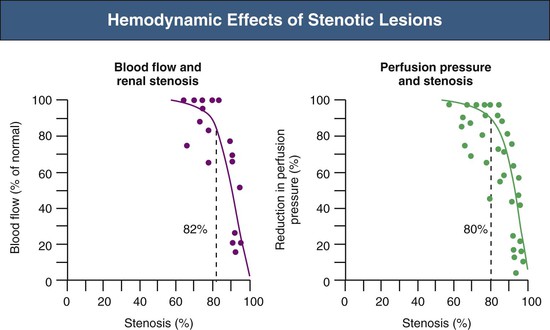
Renovascular occlusive disease from any cause can activate pressor pathways that tend to restore renal artery perfusion pressures. Most notable among these is activation of the renin-angiotensin aldosterone (RAAS) system. Activation of plasma renin only occurs after poststenotic pressures fall by at least 10% to 20% compared with aortic pressures.2 A fall in renal perfusion pressure sufficient to initiate RVH only occurs when luminal occlusion is relatively severe, usually in the 70% to 80% cross-sectional occlusion range (Fig. 39-1, left).
When critical stenosis develops and reduces renal perfusion pressure, multiple mechanisms are activated in the kidney to restore renal blood flow. Central to this process is the release of renin from the juxtaglomerular apparatus, leading to activation of the RAAS. This is mediated in part by stimulation of neuronal nitric oxide synthase and cyclooxygenase-2 in the macula densa. Blockade of the RAAS during creation of an experimental RA lesion prevents development of hypertension. Studies in transgenic mice without receptors to angiotensin confirm that development of RVH requires an intact RAAS.3 In the absence of RAAS blockade, systemic arterial pressures increase until renal perfusion is restored. Studies in both experimental models and humans indicate that additional mechanisms contribute to long-term BP elevation in the presence of RA stenosis, including activation of the sympathetic nervous system, impairment of nitric oxide generation, and release of endothelin, as well as hypertensive microvascular injury to the nonstenosed kidney.4
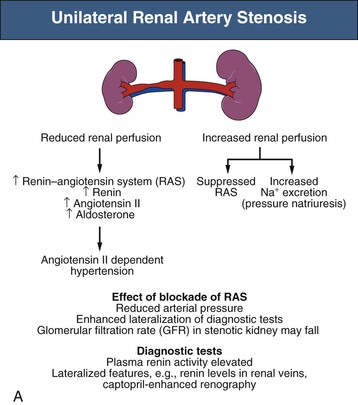
Mechanisms responsible for sustained RVH differ depending on whether one or both kidneys are affected by significant stenoses, either pathologic or created in animal models using clips (Fig. 39-2). The nomenclature distinguishes between a situation in which one clip is present with a normal contralateral or unclipped kidney (“1-clip–2-kidney hypertension”) and a situation in which the entire renal mass is affected with no contralateral kidney (“1-clip–1-kidney hypertension”). Both these situations depend on impaired renal perfusion and initial activation of the RAAS with sodium retention. However, the presence of a normal contralateral kidney allows pressure natriuresis to occur, in which the elevated perfusion pressure mediates natriuresis in the nonstenotic kidney. Because the nonstenotic kidney functions to eliminate the excess sodium, the level of perfusion to the stenotic side remains reduced, leading to sustained activation of the RA stenosis. This sequence of events produces angiotensin II (Ang II)–dependent hypertension and secondary aldosterone excess with hypokalemia (Fig. 39-2, A).
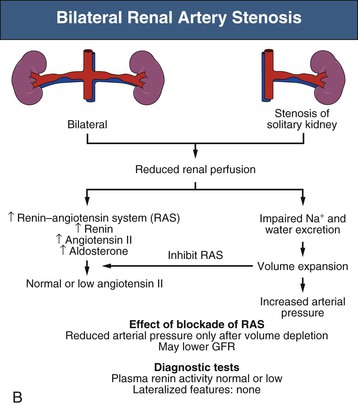
By contrast, 1-clip–1-kidney hypertension represents a model in which the entire renal mass is exposed to reduced pressures beyond a stenosis. There is no normal or nonstenotic kidney to counteract increased systemic pressures. As a result, sodium is retained and blood volume expanded, which eventually feeds back to inhibit the RAAS (Fig. 39-2, B). Therefore, 1-clip–1-kidney hypertension is typically not angiotensin dependent unless removal of volume is achieved that reduces renal perfusion pressure and activates the RAAS.
These differences have clinical implications. Many diagnostic studies used to evaluate the functional significance of RA lesions depend on comparisons of the different physiologic response of the two kidneys, which may give a false impression if both kidneys are involved. Furthermore, diagnostic tests that depend on differences in responses to alterations in sodium status (e.g., measuring renal vein renin levels after sodium depletion or individual kidney sodium reabsorption) may be problematic, because high levels of Ang II and aldosterone stimulate sodium reabsorption in both the stenotic and the nonstenotic kidney. This partly accounts for the less frequent use of such tests in recent years.5
Variations in the natural history of RVH complicate our understanding of primary pathogenic mechanisms. Rarely is it known exactly when critical levels of stenosis develop. In experimental models, the relative importance of pressor mechanisms, including measurable activation of the RAAS, changes with time. Levels of circulating plasma renin activity decrease, as does the responsiveness of BP to short-term blockade of the renin system. Several mechanisms have been proposed to explain such changes, including a slowly developing pressor action of Ang II, a transition to alternate pressor mechanisms, and intrinsic renal injury to the nonstenotic kidney, which ultimately sustains hypertension despite reversal of the vascular lesion. In experimental models, this translates into a time limit for reversibility of RVH.
There are several clinical correlates with this variable time course. First, it is not known how best to identify when revascularization will fail to improve BP control, although a brief duration of hypertension suggests a more favorable response to intervention. As a result, many of the diagnostic studies that depend on lateralization of effects have only modest predictive value when negative. As a general rule, studies are most reliable when positive, meaning that high-grade lateralization accurately predicts an improvement after revascularization. Second, coexistent intrarenal disease, such as arteriolosclerosis with glomerulosclerosis, is associated with persistent hypertension despite correction of RA stenosis, particularly for patients with ASRVD.6 In these patients, a long duration of hypertension allows the development of arteriolosclerotic lesions and renal injury in the contralateral kidney (Fig. 39-2, A). Thus, older age and a long duration of hypertension (e.g., >3 to 5 years) predict a poorer outcome of intervention in this patient population. Most elderly patients with ASRVD also have impaired renal function related to microvascular renal injury in addition to main RA stenosis.
Atherosclerotic Renovascular Disease
Epidemiology
Atherosclerotic narrowing of the renal arteries generally occurs in older patients (>50 years) and is associated with systemic atherosclerosis. Younger patients with premature atherosclerosis are also at risk. ASRVD is the most common cause of RVH and can contribute to loss of renal function, leading to end-stage renal disease (ESRD) (Fig. 39-3). Atherosclerotic plaque often arises in the first or second centimeter of the renal artery or may extend from the aorta into the renal ostium. Aortic and renal vascular calcification is often present. ASRVD is a manifestation of systemic atherosclerosis and is associated with coronary, cerebrovascular, peripheral vascular, and aortic disease.7,8 The prevalence of ASRVD appears to be increasing. Although this partly reflects improved imaging and selection bias, this trend likely also reflects that more people are surviving to ages when atherosclerotic vascular disease in the visceral abdominal vessels reaches critical levels, producing RVH when the kidney is affected. In patients undergoing angiography of the peripheral or coronary circulation, ASRVD is found in 11% to 42%.9 Predictors of ASRVD include a history of hypertension, presence of renal functional impairment, coexisting vascular or coronary artery disease, the presence of abdominal bruits, and a history of smoking. RA lesions are bilateral in 20% to 40% of such patients.
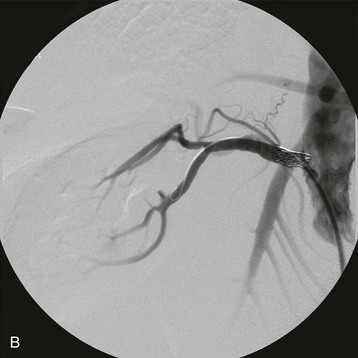
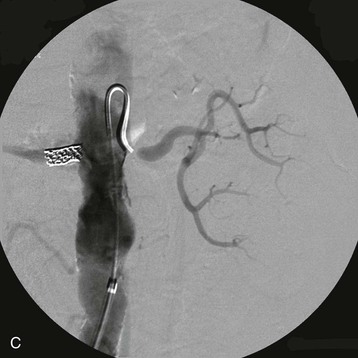
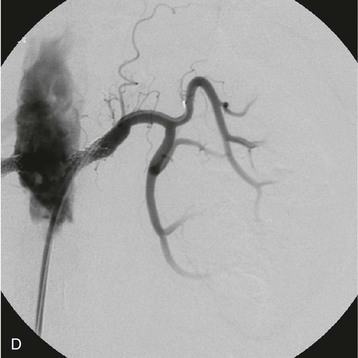
Estimates of the prevalence of ASRVD depend on the population screened. One population-based study of 870 patients older than 65 screened with RA duplex ultrasound found a 6.8% prevalence of ASRVD, defined as greater than 60% stenosis. No differences were detected between African Americans and Caucasians.10 Autopsy series report an overall prevalence of 4% to 20%, with progressively higher rates for those older than 60 years (25% to 30%) and 75 years (40% to 60%). These studies suggest that ASRVD is common among elderly hypertensive patients. Furthermore, RA stenosis from ASRVD has been reported to contribute to the decline in renal function in 15% to 22% of patients reaching ESRD.11,12
Relationship to “Ischemic” Renal Disease
Activation of pressor mechanisms producing RVH can occur without loss of kidney size or function. However, the more common clinical scenario in patients with ASRVD involves both increasing severity of hypertension and deteriorating renal function, often with loss of kidney volume. Because mechanisms underlying parenchymal renal damage differ from those responsible for generating hypertension, improved BP control after revascularization in some cases may be achieved without improvement in kidney function.
Transition from “reversible” loss of function to “irreversible” tissue fibrosis is not well understood. Basal renal energy requirements are met with less than 10% of blood flow, consistent with its filtration function. Studies using blood oxygen level–dependent (BOLD) magnetic resonance imaging (MRI) indicate that despite reductions in blood flow and GFR, many patients maintain normal cortical and medullary tissue oxygenation.13 Thus, many poststenotic kidneys have no more “ischemia” than normal kidneys. These observations explain the relative stability and infrequent progression of renal injury in prospective trials of medically treated patients with ASRVD, such as Angioplasty and Stenting for Renal Atherosclerotic Lesions (ASTRAL).14,15 Nonetheless, reduced renal perfusion ultimately activates numerous mechanisms of tissue injury. Figure 39-4 summarizes the activation of vasoactive and inflammatory pathways.16 Over time, inflammation, fibrosis, and microvascular rarefaction occur, leading to irreversible changes.16,17
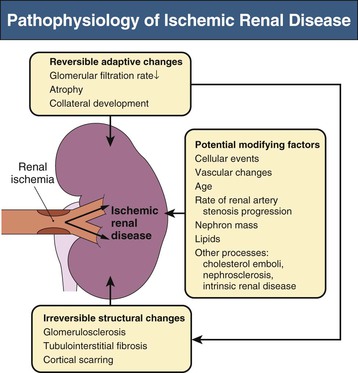
Activation of the RAAS and endothelial systems such as endothelin and oxidative stress pathways have been demonstrated with RA stenosis models.16 These systems stimulate inflammation and drive fibrosis. Under acute conditions of reduced blood flow with persistent filtration and tubular function, levels of deoxygenated hemoglobin increase in the renal medulla, representing medullary hypoxia.18 Medullary oxygen levels are normally lower than cortical levels and are heavily dependent on the level of solute transport. Reductions in GFR and associated energy-dependent solute transport allow “adaptation” to reduced blood flow without development of tissue hypoxia. Only when more severe vascular occlusion develops beyond the limits of such adaptation can one identify overt cortical ischemia associated with increased deoxyhemoglobin.19 This results in macrophage accumulation with progressive tubular cell loss and fibrosis.20 Glomeruli are usually preserved, although collapsed.
Clinical Manifestations
Renovascular Hypertension
Clinical studies suggest that for any level of BP, patients with RVH have higher nocturnal pressures (“nondipper”) and have more severe target organ manifestations, including left ventricular hypertrophy, than patients with essential hypertension21 (Box 39-2). Likelihood of RVH in patients with treatment-resistant hypertension increases with elevated cholesterol, impaired renal function, lower body mass index, and smoking; but none of these features is sufficiently sensitive or specific to offer diagnostic precision. Some reports indicate that RVH rarely may be associated with nephrotic-range proteinuria, which can regress with correction of the vascular lesions.22
Blood pressure elevations from RVH vary widely. Acute RA occlusion may only gradually produce BP increase or may cause a rapid increase in hypertension that precipitates a hypertensive urgency or emergency (see Chapter 37). Before the current era of antihypertensive agents, 30% of Caucasian patients appearing in an emergency department with hypertensive urgency (defined as grade III or IV hypertensive retinopathy) were ultimately found to have RVH. Syndromes of polydipsia and accelerated hypertension with hyponatremia and hypokalemia, sometimes attributed to the dipsogenic actions of Ang II, also have been observed.
Recent consensus documents emphasize the need for effective population-wide BP control, while limiting the number and expense of diagnostic studies. As a result, most patients with hypertension simply are treated and subjected to few laboratory investigations. For those who reach acceptable BP control without adverse effects, no further studies are performed. Therefore, many if not most cases of RVH are not detected (Fig. 39-5), unless hypertension becomes more difficult to treat or renal dysfunction ensues.
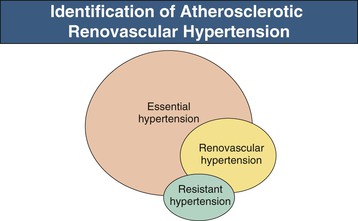
In recent years, widespread application of RAAS blockers for indications other than hypertension, including congestive cardiac failure, diabetic nephropathy, and other proteinuric renal disease, increases the exposure of individuals with undetected RA stenosis to these drugs.23
A consequence of these changes has been the emergence of distinctive clinical syndromes that merit evaluation in patients at risk for ASRVD (see Box 39-2). As a result, patients that typically undergo diagnostic evaluation and renal revascularization are a subset of the patient population with RVH. This subset is characterized generally by more severe hypertension, decreasing renal function, propensity for rapid volume accumulation manifested as “flash” pulmonary edema, and occasionally advanced renal failure.
Ischemic Renal Disease
The diagnosis of IRD (ischemic nephropathy) should be considered in several clinical settings (see Box 39-2).
Renal Impairment in Patients with Renovascular Hypertension or in Atherosclerotic Age Range
Ischemic nephropathy should be considered as a cause of chronic kidney disease (CKD) in the atherosclerotic age group, particularly when other vascular disease is detected. Many but not all will have a history of hypertension. It remains difficult to separate primary vascular disease from renal parenchymal injury associated with nephrosclerosis from other vascular insults. Clues to IRD include asymmetry of kidney size or recent deterioration of renal function, as opposed to slowly progressive CKD. An asymmetrically small kidney in an adult over age 50 has a 70% chance of being associated with ipsilateral RA stenosis.24
The development of renal impairment in patients treated medically for longstanding hypertension should raise suspicion for possible IRD. Given the high frequency of bilateral RA stenosis (30% to 50%), the possibility of IRD should always be considered in patients with known prior RA stenosis. The most common presentation of ASRVD is unilateral involvement in a patient with CKD associated with longstanding hypertension. In this case, renal impairment cannot be attributed solely to IRD because the RA stenosis affects only one kidney. The kidney supplied by the stenotic vessel may have reduced blood flow, resulting in diminished GFR, RVH, and atrophy. The contralateral kidney with a patent renal vessel often hypertrophies and compensates with hyperfiltration. However, over time, this kidney develops parenchymal injury. In some cases, the kidney with the patent RA has worse renal function than the poststenotic kidney.25
Acute Kidney Injury After Starting Antihypertensives/RAAS Blockade
Patients with hemodynamically significant RA stenosis to the entire functioning nephron mass are at risk for the development of “functional” acute kidney injury (AKI) after starting RAAS blockers. The sudden reduction in systemic BP in the patient with critical RA stenosis may reduce RA pressure below levels needed to sustain glomerular filtration by autoregulation (see Medical Therapy). This can occur with reduction in BP by any antihypertensive agent. With medications that block the production or action of Ang II, alterations in glomerular hemodynamics may result in acute reduction in GFR, which is independent of effects on systemic BP.26 Normally, activation of Ang II causes efferent arteriolar vasoconstriction, which preserves transcapillary filtration pressures at the glomerulus when preglomerular pressures are reduced, thereby maintaining GFR. The loss of this compensatory mechanism induced by agents that inhibit or block the RAAS can result in functional AKI. This typically occurs within a few days from the start of therapy and is usually, but not always, reversible.
In patients without RA stenosis, AKI can also occur with the use of RAAS inhibitors. This most frequently occurs in patients with cardiac or hepatic dysfunction or patients with intravascular volume depletion because, in these settings, GFR is also Ang II dependent. In a prospective study, the observation of 20% increase in serum creatinine or more after administration of an angiotensin-converting enzyme (ACE) inhibitor detected most cases of bilateral severe RA stenosis.27 When prospectively challenged, more than 90% of a cohort followed in the United Kingdom tolerated RAAS blockade, including 78% of those with high-grade, bilateral ASRVD.28 Because of the potential change in GFR, kidney function should be rechecked within 1 to 2 weeks of instituting therapy with RAAS blockers.29
“Flash” Pulmonary Edema
Some patients with bilateral RA stenosis or ischemic nephropathy develop severe hypertension and extracellular fluid volume excess caused by impaired pressure natriuresis. Such conditions may produce the sudden (“flash”) onset of pulmonary edema in association with rapid development of circulatory congestion.30 This has been attributed in part to rapid loss of contractile strength of the left ventricle caused by sudden increases in afterload. Impaired diuresis can result in exaggerated swings in BP, circulatory congestion, and renal function.31 Such patients have increased mortality and hospitalization rates compared with those who have congestive heart failure (CHF) without renovascular disease.32 Case series suggest that renal revascularization can facilitate fluid volume management, reduce hospitalization, and occasionally improve cardiac function independent of interventional procedures for the heart itself, even in patients with poor preoperative cardiopulmonary status.33,34 In one series, 41% of patients with bilateral RA stenosis had a history of pulmonary edema, compared with 12% with unilateral renovascular disease. Of patients with bilateral RA stenosis, 77% had no further pulmonary edema after RA stent placement in one or both arteries. The patients who did have recurrent pulmonary edema all had evidence of stent thrombosis or restenosis.35
No prospective clinical studies have examined the effect of optimal medical therapy of BP and volume status on the frequency of events in these patients. Observational, retrospective studies indicate that ASRVD in patients with CHF imparts a worse survival and that renal revascularization lowers the rates of recurrent pulmonary edema and hospitalization as compared to those treated medically.32 Reports from prospective, randomized controlled clinical studies such as ASTRAL, however, demonstrate no differences in hospitalizations or episodes of CHF between intensively treated patients with or without RA stenting.14 An important caveat related to this observation is that patients considered “likely to benefit” from revascularization were excluded from ASTRAL.36
Oligoanuric Acute Superimposed on Chronic Kidney Disease
Patients with high-grade RA stenosis are at risk for renal artery occlusion. When RA stenosis is bilateral or unilateral in the patient with a single functioning kidney, progression to total occlusion can present as oligoanuric AKI, sometimes associated with a hypertensive emergency. A clinical clue to this diagnosis is abrupt onset oligoanuria. In this setting, the kidney parenchyma may be viable despite lack of filtration; in some patients collateral vessels maintain renal viability in the face of proximal RA occlusion. Clues to renal viability include preserved kidney length and evidence of renal contrast enhancement (“renal blush”) seen on delayed or venous phase images during renal angiography. When these factors are present and the clinical course is consistent with recent occlusion, there is a chance of retrieval of renal function if revascularization is feasible clinically and anatomically. In this setting, urgent vascular surgical consultation should be sought, and the nephrologist should assume that the kidneys may be viable for weeks to months.
Incidental Renal Artery Stenosis
As mentioned, ASRVD is highly correlated with disease in both the coronary and the peripheral vasculature. Disease is usually identified incidentally at angiography or computed tomography (CT) for other indications. Most of these patients with coexisting ASRVD and coronary artery disease have only moderate degrees of RA stenosis (50% to 75%) with minimal hemodynamic impact. However, the presence of RA lesions is an independent risk factor for mortality, which may reach 30% over 4 years in high-risk groups.37 Since no data support intervention for asymptomatic RA stenosis, the use of screening renal aortography at coronary angiography should be limited to patients who have demonstrated clinical manifestations, as summarized earlier, and in whom documentation of RA disease is likely to influence management.
Natural History
Early, retrospective angiographic studies (1980s) suggested ASRVD typically progresses over 2 to 5 years. “Progression” is usually defined as a greater than 25% luminal diameter narrowing or as severe stenosis progressing to vascular occlusion. Over 4 to 5 years, 6% to 16% of stenoses advance to occlusion. Prospective studies between 1990 and 1997 using Doppler ultrasound in patients with atherosclerotic RA lesions indicated hemodynamic progression in 30% over 3 years, varying by degree of initial stenosis, although total occlusion occurred in only 3%.38 Measurable loss of kidney length (>1 cm) is less common but accompanies progressive vascular occlusion. Changes in serum creatinine are often minimal.39
Progression is most likely in patients with more than 60% stenosis. It often occurs without changes in BP control. Therapy with statins appears to reduce the risk of progression and occasionally induces regression of atherosclerotic RA stenosis.40 Statins also appear to attenuate renal parenchymal injury associated with experimental ASRVD.41
Follow-up studies of patients with incidentally detected, high-grade RA stenosis (>70%) treated medically indicate that fewer than 10% required later revascularization for intractable hypertension.42 Another report noted that few patients with incidental RA stenosis progressed to ESRD over follow-up of 8 to 9 years.43 Patients followed after simultaneous renal angiography at cardiac catheterization noted no difference in serum creatinine between those with and without RA stenosis at follow-up.
Risk of Mortality
Both ASRVD and ischemic nephropathy (IRD) are associated with limited long-term survival, consistent with widespread atherosclerotic disease. Retrospective analyses report 3-year to 5-year mortality rates of 30% to 35% in patients with RA stenosis, largely caused by cardiovascular events or cerebrovascular accident (stroke). In follow-up of more than 1200 patients who underwent coronary and renal angiography, patients with RA stenosis had a 65% 4-year survival versus 85% for those without RA stenosis at catheterization. The 5- and 10-year survival rates for patients reaching ESRD caused by IRD are as low as 18% and 5%, respectively.44
Whether renal revascularization improves overall survival in patients with ASRVD remains controversial.45 In one study, patients with bilateral ASRVD who were treated medically had better follow-up renal function than those who had undergone revascularization, but there was no difference in survival.46
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


