pathogenesis, those against dsDNA and those against the complement component C1q.
Circulating levels of C3 and C4 are lower in active LN compared to inactive LN or nonrenal SLE, indicating ongoing complement activation.30,31
Longitudinal assessment of circulating C3 and C4 levels during SLE flare showed that levels decrease significantly at the time of a renal flare, but not at nonrenal flare, even if the nonrenal flare occurred in patients with a history of LN.29
Renal tubular production of C3 and complement factor B occurs in LN patients but not healthy controls.34,35
The inflammatory receptor for C3a (C3aR), absent from healthy kidneys, becomes expressed in glomerular endothelium in association with IC deposits in LN, and the expression level correlates with LN severity.36
The inflammatory receptor for C5a (C5aR), although present in normal kidneys, is greatly upregulated in the mesangium and podocytes of LN kidneys.37
The expression of CR1 is decreased in LN glomeruli, compared to its normal expression on podocytes.38
The expression of another complement regulator, decay accelerating factor (DAF, CD55), is also reduced in LN patients from its normal expression in the juxtaglomerular apparatus, and appears de novo in the renal vasculature, interstitium, and mesangium.39
Some of these may specifically mediate kidney damage (e.g., MCP-1 and TNF-α), whereas others may predispose to kidney injury through general effects on autoimmunity.
genes as susceptibility genes for SLE onset,79 an increase in IFN-α induced gene expression (the IFN-α signature) associated with active SLE,92 and the number of known SLE autoantigens that can drive IFN-α secretion.
and environmental factors interact and contribute to LN become clearer, so too will our understanding of the pathogenesis of LN.
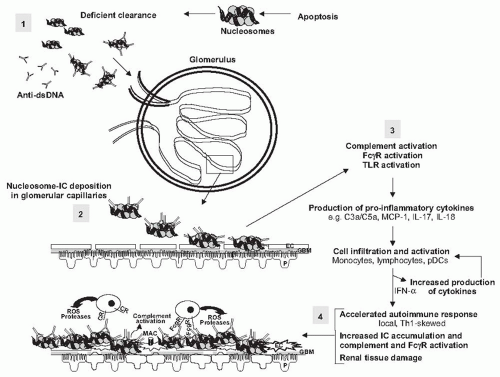 FIGURE 53.1 A paradigm for lupus nephritis pathogenesis. The onset of lupus nephritis likely begins with initial accumulation of self-antigen and immune complexes. In the model presented in the figure, the self-antigens are nucleosomes that can persist due to deficient clearance or overwhelming production, and which in turn can drive anti-dsDNA production (step 1, in shaded box). The resulting immune complexes (IC) are prone to deposit in the renal vascular beds (step 2), in part due to the positively charged core histones of the nucleosome. Once deposited, the IC can activate the complement system, activate circulatory leukocytes via expressed FcγR, and activate resident cells expressing TLRs (step 3). This establishes a cascade of inflammatory cytokine and chemokine production that recruits and activates inflammatory cells, lymphocytes, and pDCs. These infiltrating cells further amplify the production of cytokines and chemokines in the kidney microenvironment. The result is a locally driven and accelerated autoimmune response with Th1 characteristics, and increased IC accumulation and accompanying complement and FcγR activation. This response culminates in the production of inflammatory mediators of tissue damage (step 4). One immediate consequence is destruction of the glomerular filtration barrier through the damaging effects on glomerular endothelial cells (EC), glomerular basement membrane (GBM), and podocytes (P), which leads to proteinuria and hematuria, the hallmark clinical manifestations of active lupus nephritis. CR, complement receptor; MAC, complement membrane attack complex. |
of infection, medications, nephrotoxins, hemolysis, thrombosis, and cardiac failure.
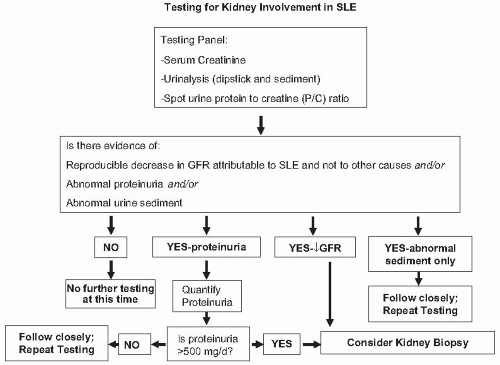 FIGURE 53.2 An algorithm for the evaluation of the kidney in patients with systemic lupus nephritis. Note that patients with a history of lupus nephritis and previous kidney biopsy may not need a repeat biopsy (see text). Kidney biopsy should be done for all new diagnoses of kidney involvement. |
Not all kidney disease in SLE patients is classic, IC-mediated glomerulonephritis (LN), so one therapy does not fit all patients. For example non-LN glomerular diseases have been reported in SLE patients.119,120,121 This literature is mostly case reports, but in a series of 252 patients, 5% were found to have changes consistent
with focal segmental glomerulosclerosis, minimal change disease, thin glomerular basement membrane disease, hypertensive nephrosclerosis, and amyloidosis.119 The incidence of podocytopathies in lupus patients appears to be greater than in the general population, suggesting a causal link to the immune dysregulation of SLE.122,123 Amyloid A (AA) amyloidosis has also been reported frequently in some series.120,121 Finally, there are other important kidney lesions found in SLE patients that are treated differently than LN, such as interstitial nephritis without glomerulonephritis121 and thrombotic microangiopathy with or without LN.124,125
The kidney biopsy, especially if performed serially, assesses the degree of chronic kidney injury, and therefore the risk of progressive renal failure that is not related to active LN. If extensive scarring is the dominant process found on biopsy even with some areas of active inflammation, the risk of immunosuppression may outweigh its benefits in terms of renal survival. Such patients may be more appropriately treated with kidney-protective therapies alone.
In the context of LN therapeutics, kidney biopsies can and should be exploited in novel ways to better inform future drug development. For example, leukocyte subsets can be analyzed by specific staining in lupus kidneys and may yield new insights on renal inflammation.126 Proteomic techniques can be used to look for patterns of protein expression in LN.127,128 Gene expression in biopsies can be analyzed with microarray techniques.128,129 These technologies are just being applied to kidney biopsies, but have the potential to greatly enhance the amount of information available from renal tissue.
Mesangial hypercellularity. Mesangial hypercellularity is almost always present in LN, except in Class I (Fig. 53.3), and is the basic, and probably the earliest LN lesion which is later combined with other pathologic patterns of injury.
Endocapillary hypercellularity. Endocapillary hypercellularity is the hallmark lesion in forms of proliferative LN (Figs. 53.4 and 53.5). Intracapillary cells usually are infiltrating inflammatory cells (including monocytes/macrophages, polymorphonuclear leukocytes, lymphocytes, and rarely eosinophils or basophils). There may also be a component of endothelial cell proliferation.
Extracapillary hypercellularity. Extracapillary proliferation results in crescent formation (Fig. 53.6), and is common in proliferative forms of LN. It is frequently associated with glomerular capillary rupture, Bowman’s capsular basement membrane rupture, fibrin in Bowman’s space, and fibrinoid necrosis of the glomerular capillary tuft.
Karyorrhexis with or without associated fibrinoid necrosis of the glomerular capillary tuft (Figs. 53.7 and 53.8). Karyorrhexis in glomeruli usually reflects apoptosis, a common finding in LN. The apoptotic cells may be infiltrating inflammatory cells or native glomerular cells. Hematoxylin bodies (Fig. 53.9), seen occasionally in
biopsies, most likely represent a tissue equivalent of the LE cell phenomenon.
Wire loop lesions. These lesions are due to large subendothelial immune complex deposits, visible even with light microscopy (Fig. 53.10). If these subendothelial deposits are large enough, they may occlude the entire glomerular capillary lumen and appear as “hyalin thrombi” (Figs. 53.7 and 53.10). Wire loop lesions are positive for periodic acid-Schiff (PAS), negative with methenamine silver stain, and red with Masson’s trichrome stain. Wire loop lesions are much more common in LN with global glomerular hypercellularity than with biopsies showing mainly segmental hypercellularity and/or necrosis.
Spikes. Diffuse uniform glomerular capillary loop thickening with “spike” formation on methenamine silver stain (Figs. 53.11 and 53.12) is the main light microscopic pattern of injury if the immune complex deposits are subepithelial in membranous lupus nephritis.
TABLE 53.1 Classification of Lupus Nephritis | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
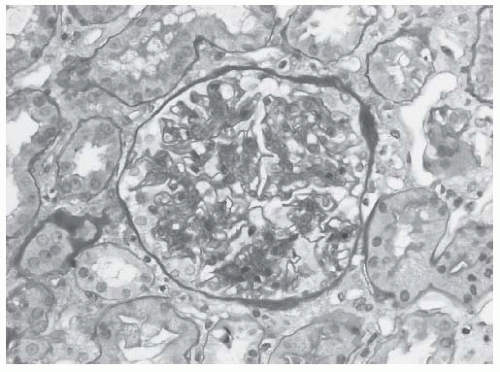 FIGURE 53.3 Mesangial hypercellularity in a case of class II lupus nephritis. Note that the glomerular capillaries are patent. (Periodic acid-Schiff [PAS] X400.) |
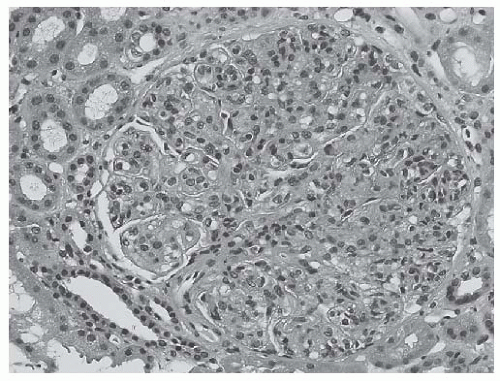 FIGURE 53.4 Global endocapillary hypercellularity with obliteration of the glomerular capillaries in a case of class IV lupus nephritis. The hypercellularity is the result of infiltrating inflammatory cells, including occasional polymorphonuclear leukocytes, as well as proliferating glomerular cells, including endothelial cells and mesangial cells. (Hematoxylin and eosin [H&E] X400.) |
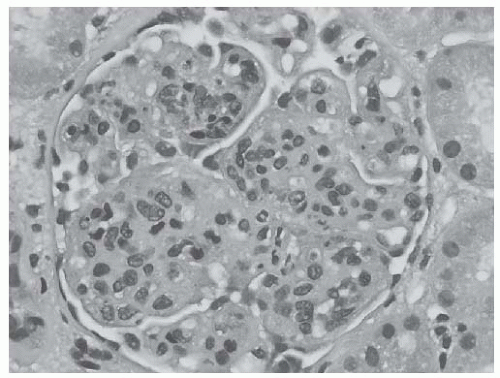 FIGURE 53.5 Global endocapillary hypercellularity with accented lobularization of the glomerular capillary tuft, resembling a membranoproliferative glomerulonephritis in a case of class IV lupus nephritis. (H&E, X400.) |
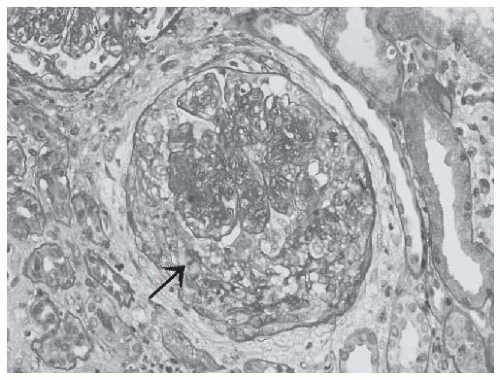 FIGURE 53.6 A cellular crescent in a case of class IV lupus nephritis. Note the compressed glomerular capillary tuft and the rupture in the Bowman’s capsule (arrow). (PAS X400.) |
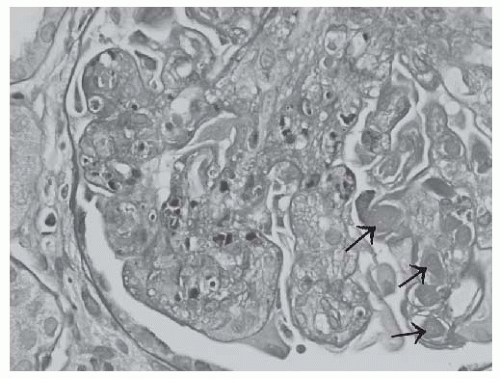 FIGURE 53.7 Apoptotic debris (karyorrhectic nuclei) in the glomerular capillaries in a case of class IV lupus nephritis. In this glomerulus, large subendothelial deposits (“wire loop” lesions) and intracapillary hyalin thrombi (arrows) are also present. (PAS X600.) |
differentiates active (A) and chronic (C), and segmental (S) and global (G) glomerular lesions. Active glomerular lesions include glomerular endocapillary hypercellularity with or without leukocyte infiltration and with substantial luminal reduction, karyorrhexis, fibrinoid necrosis, rupture of the glomerular basement membrane, cellular or fibrocellular crescents, wire loop lesions, and large intraluminal immune complexes (hyalin thrombi) (Figs. 53.4,53.5,53.6,53.7,53.8 and 53.10). Chronic lesions include glomerular sclerosis (segmental or global), fibrous adhesions, and fibrous crescents (Figs. 53.13,53.14,53.15). Segmental lesions involve less than half of the glomerular capillary tuft area; global lesions involve more than 50% of the glomerular capillary tuft area.
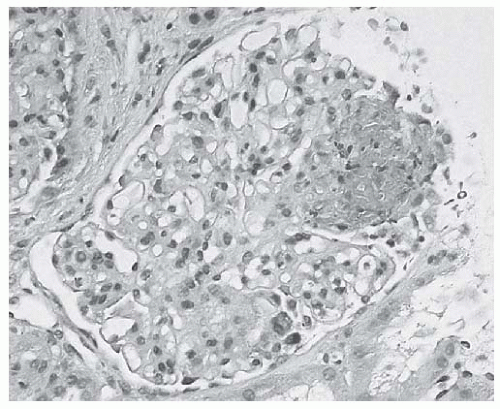 FIGURE 53.8 Segmental glomerular capillary tuft necrosis associated with karyorrhectic/apoptotic debris in a case of focal lupus nephritis. (H&E X400.) |
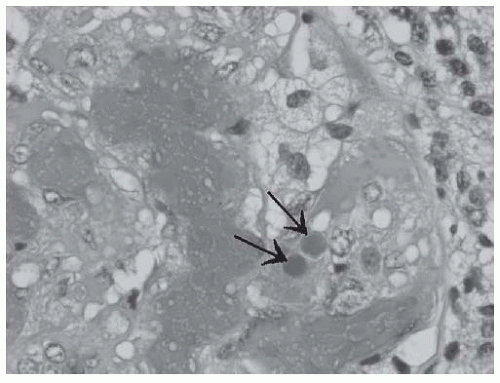 FIGURE 53.9 Hematoxylin bodies in a glomerular capillary (arrows) in a case of active class IV lupus nephritis. (H&E X1000.) |
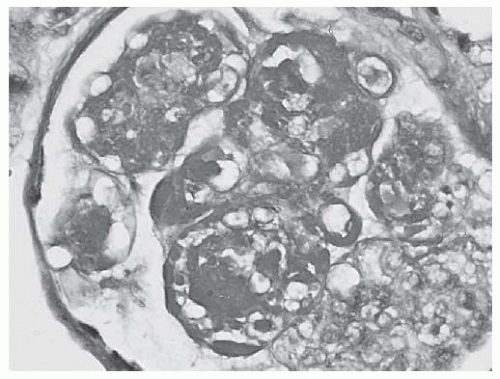 FIGURE 53.10 Large PAS positive deposits along the glomerular capillary loops (“wire loop” lesions) as well as extensive mesangial deposits and glomerular capillary hyalin thrombi in a case of class IV lupus nephritis. (PAS X600.) |
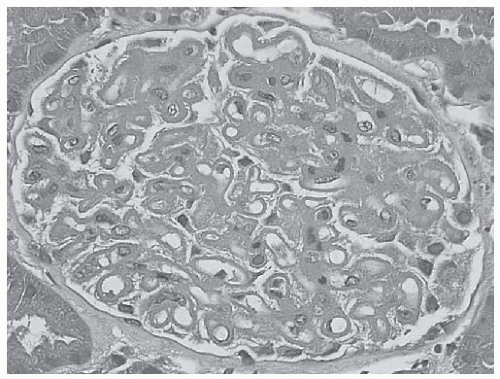 FIGURE 53.11 Diffuse uniform glomerular capillary thickening without hypercellularity in a case of membranous class V lupus nephritis. (H&E X400.) |
glomerular capillary immune complex deposits are present, the diagnosis of class II LN should not be made.
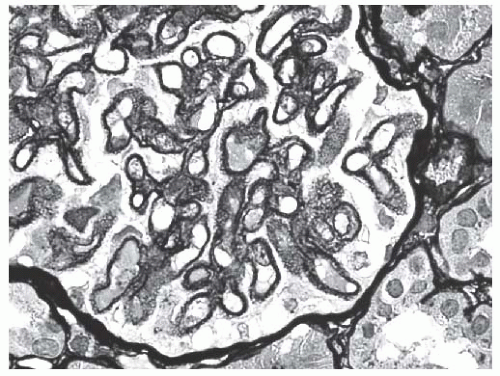 FIGURE 53.12 Methenamine silver stain reveals extensive spike formation along the glomerular capillary loops in the same biopsy shown in Figure 53.11. (Jones’ methenamine silver X600.) |
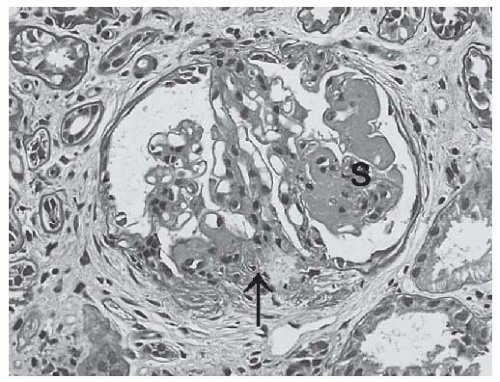 FIGURE 53.13 Segmental sclerosis (S) and glomerular capillary adhesion (arrow) in a glomerulus from a biopsy with class III lupus nephritis. (PAS X400.) |
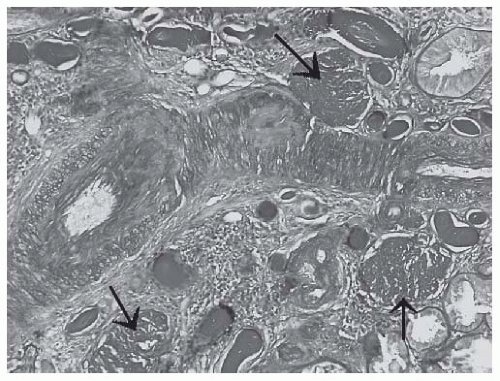 FIGURE 53.14 Globally sclerotic glomeruli (arrows) in a biopsy with advanced sclerosing (class VI) lupus nephritis. (PAS X200.) |
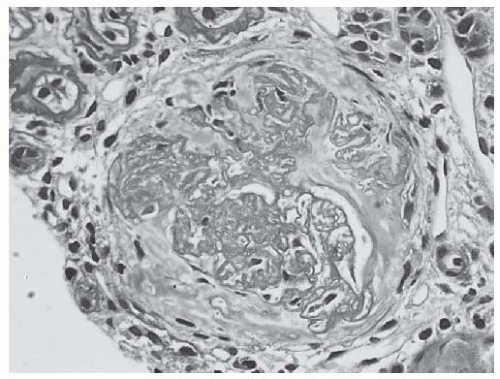 FIGURE 53.15 A fibrous crescent from biopsy with class IV lupus nephritis with moderate to advanced chronicity and mild activity. Note the disrupted Bowman’s capsule and the separation of sclerosing glomerular lobules by faintly PAS positive interstitial type collagen. (PAS X400.) |
In class III (A) there are only active lesions (focal proliferative LN).
In class III (A/C) both active and chronic lesions are present (focal proliferative and sclerosing LN). In such cases, focal or segmental sclerosing glomeruli coexist with glomeruli with active proliferative/necrotizing lesions.
In class III (C) only focal sclerosing glomerular lesions are noted with glomerular scars and segmental or global sclerosis (focal sclerosing LN). Active lesions are not seen.
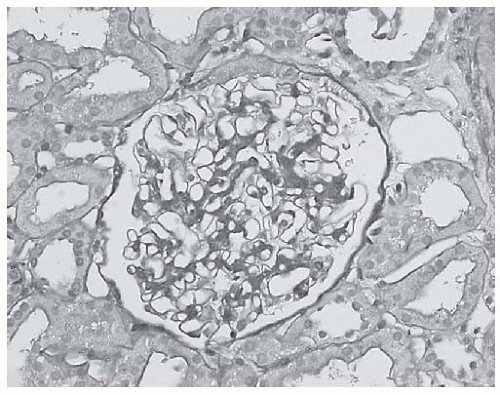 FIGURE 53.16 A light microscopically unremarkable glomerulus in a biopsy with class I lupus nephritis. Immunofluorescence and electron microscopy revealed mesangial immune complex deposits. (PAS X400.) |
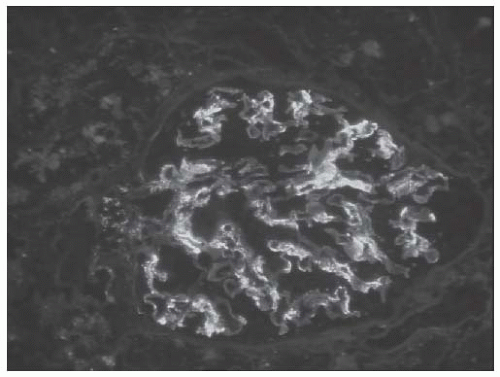 FIGURE 53.17 Mesangial immune complex deposits in a case of class II lupus nephritis. (Direct immunofluorescence with an antibody to IgA, X400.) |
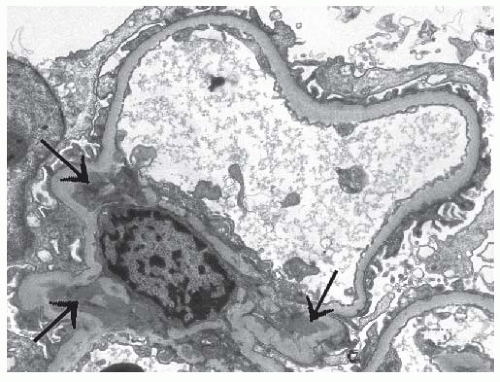 FIGURE 53.18 Mesangial electron dense immune type deposits (arrows) in a case of class II lupus nephritis. (Uranyl acetate, lead citrate X8000.) |
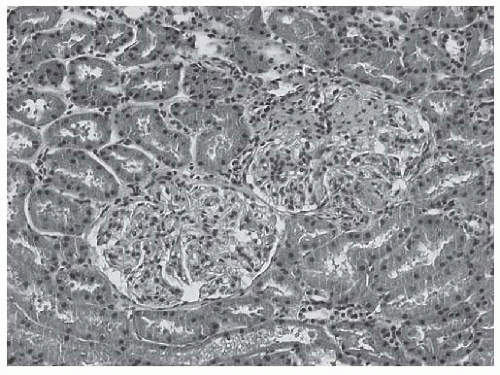 FIGURE 53.19 Two glomeruli from a biopsy with class III lupus nephritis. Note that the left lower glomerulus is light microscopically unremarkable whereas the right upper glomerulus reveals segmental proliferative lesions. (H&E, X200.) |
Class IV-S(A) indicates active diffuse segmental endocapillary or extracapillary proliferative glomerular lesion or necrosis involving more than 50% of the glomeruli.
Class IV-G(A) shows diffuse global LN with active endocapillary or extracapillary proliferative glomerular
lesions and/or necrosis involving more than 50% of glomeruli.
Class IV-S(A/C) indicates diffuse segmental proliferative and sclerosing LN. In such biopsies, active segmental proliferative lesions coexist with chronic sclerosing glomerular lesions.
Class IV-G(A/C) indicates diffuse global proliferative and sclerosing LN. These biopsies show active global proliferative lesions with chronic sclerosing glomerular lesions.
Class IV-S(C) indicates diffuse segmental sclerosing LN. In this subclass, no active lesions are present; only inactive, mainly segmental glomerular lesions are seen, such as segmental sclerosis/scarring.
Class IV-G(C) shows diffuse global sclerosing LN. In such biopsies, glomeruli reveal global sclerosis or scarring with or without fibrous crescents, involving more than 50% of all glomeruli, in the absence of active proliferative lesions.
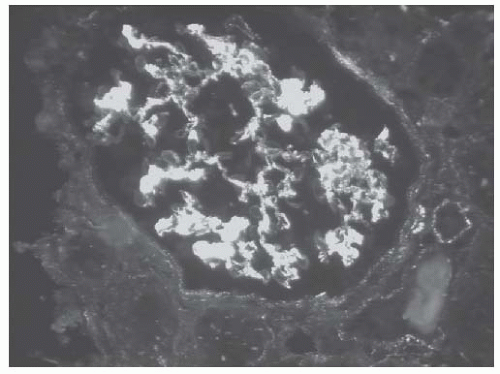 FIGURE 53.20 Granular mesangial and segmental glomerular capillary loop deposits in a case of class III lupus nephritis. Also note the subtle granular tubulointerstitial staining. (Direct immunofluorescence with an antibody to IgG, X400.) |
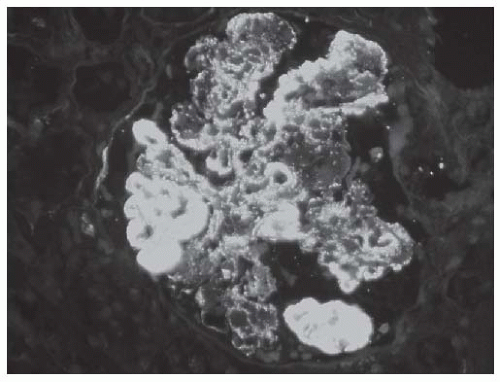 FIGURE 53.21 Diffuse granular glomerular deposits with large subendothelial deposits (“wire loop” lesions) in a case of class IV lupus nephritis. (Direct immunofluorescence with an antibody to IgG, X400.) |
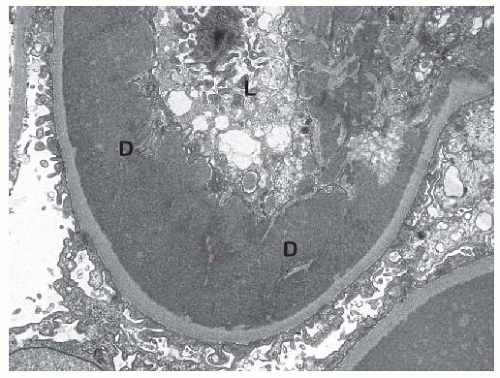 FIGURE 53.22 This electron micrograph shows a large subendothelial electron dense deposit (d) in the same biopsy shown in Figure 53.21. L, glomerular capillary lumen. (Uranyl acetate, lead citrate, X8,000.) |
sclerosis is secondary to LN. Immunofluorescence and electron microscopy still frequently reveal mild glomerular immune complex deposits in the few nonsclerotic glomeruli.
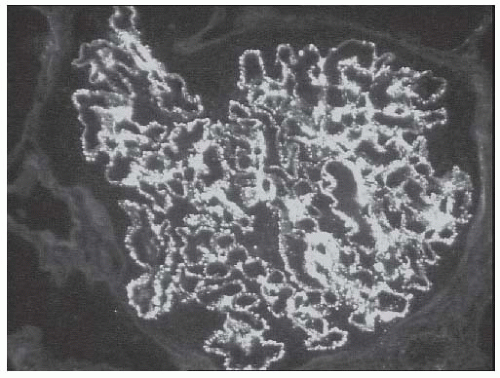 FIGURE 53.23 Granular mesangial and glomerular capillary fluorescence with an antibody to IgG in a case of membranous (class V) lupus nephritis. Note that over 50% of the glomerular capillaries contain granular deposits. (Direct immunofluorescence, X400.) |
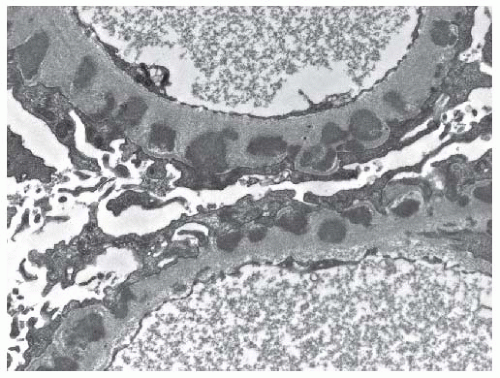 FIGURE 53.24 Subepithelial electron dense immune type deposits along the glomerular basement membrane in a case of class V (membranous) lupus nephritis. Note that occasional deposits are already completely incorporated into the glomerular basement membrane. (Uranyl acetate, lead citrate X15,000.) |
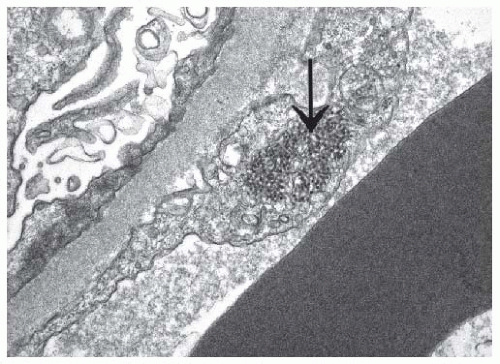 FIGURE 53.25
Get Clinical Tree app for offline access
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|





