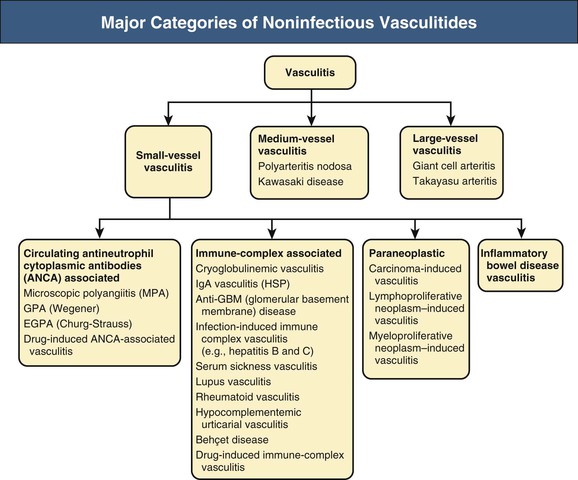
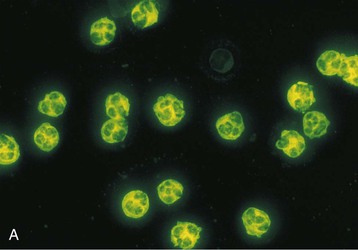
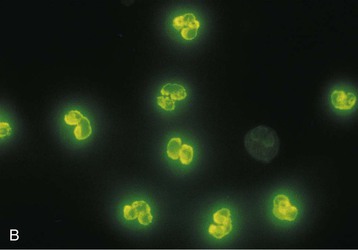
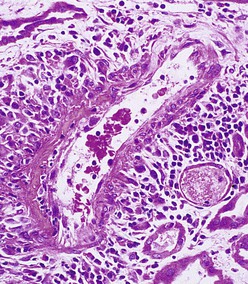
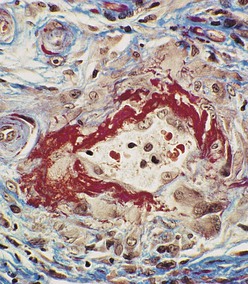
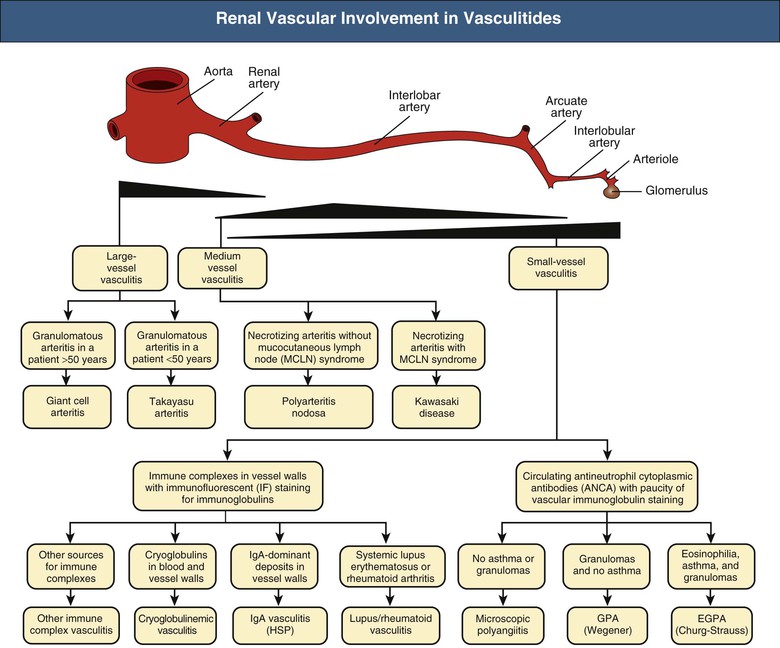
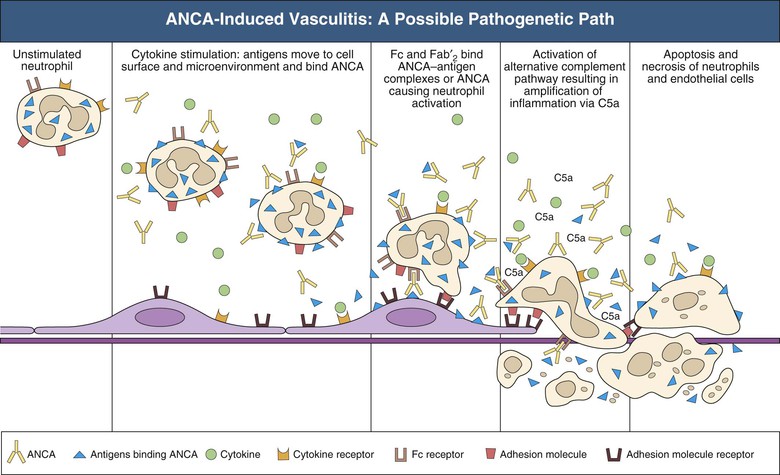
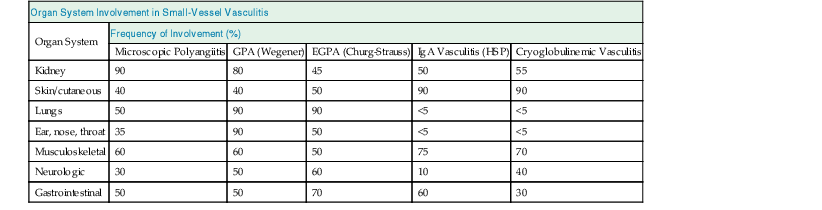
J. Charles Jennette, Ronald J. Falk, JulieAnne G. McGregor The kidneys are targets for a variety of systemic vasculitides, especially those that affect small vessels.1–5 This is not surprising given the large number and variety of renal vessels. Vasculitis involving the kidneys can produce a wide variety of clinical manifestations, depending mainly on the type of renal vessel affected. Vasculitides can be categorized as large-vessel vasculitis, medium-vessel vasculitis, and small-vessel vasculitis (Figs. 25-1 and 25-2). For the purposes of the discussion in this chapter, the 2012 Chapel Hill Consensus Conference definitions are used (Table 25-1).4 Table 25-1 Names and definitions of vasculitis. Adopted by the Chapel Hill Consensus Conference on the nomenclature of systemic vasculitis. Note that all three categories affect arteries, but only small-vessel vasculitis has a predilection for vessels smaller than arteries. ANCA, Antineutrophil cytoplasmic antibody. (Modified from reference 22.) A number of the vasculitides listed in Figure 25-2 are covered in other chapters and are not reviewed in detail here except in the context of differential diagnosis, for example, cryoglobulinemic vasculitis (Chapter 21), IgA vasculitis (Henoch-Schönlein purpura; Chapter 23), and anti–glomerular basement membrane (anti-GBM) disease (Chapter 24). Nephrologists most often encounter patients with small-vessel vasculitides because these often cause glomerulonephritis (GN). Therefore, small-vessel vasculitis is the primary focus of this chapter. Small-vessel vasculitis is necrotizing polyangiitis that affects predominantly vessels smaller than arteries, including capillaries, venules, and arterioles; however, arteries also may be involved.4 The most common renal target for small-vessel vasculitis are glomerular capillaries, and therefore the most common renal clinical manifestations are those of GN. Medium-vessel vasculitis is necrotizing arteritis that affects predominantly major visceral arteries.4 The interlobar arteries and arcuate arteries are affected most often in the kidneys, although any arteries from the main renal artery to the smallest interlobular arteries may be affected. Inflammation and necrosis of arteries may result in thrombosis or rupture, which causes renal infarction and hemorrhage, respectively. Large-vessel vasculitis is chronic granulomatous arteritis that affects the aorta and its major branches more often than other forms of vasculitis.4 When there is renal involvement, the ostia of the renal arteries and the main renal arteries are most often affected. The most common clinical renal manifestation is renovascular hypertension. Small-vessel vasculitis can be divided into immune complex small vessel vasculitis with moderate to marked vessel wall deposits of immunoglobulin and pauci-immune small-vessel vasculitis with few or no immune deposits in vessel walls.4 Pauci-immune small-vessel vasculitis often is associated with circulating antineutrophil cytoplasmic autoantibodies (ANCAs).5 The ANCA-associated vasculitides share an indistinguishable form of necrotizing small-vessel vasculitis that affects capillaries, venules, arterioles, and small arteries.1–5 Some patients with ANCA-associated vasculitis (AAV) have no evidence of involvement of arteries, even though they have involvement of glomerular capillaries, causing GN; pulmonary alveolar capillaries, causing pulmonary hemorrhage; or dermal venules, causing purpura. The clinicopathologic variants of pauci-immune small-vessel vasculitis are categorized on the basis of clinical, laboratory, and pathologic findings, as follows4: MPA, GPA, and less frequently EGPA share an indistinguishable pattern of GN that is the expression of the vasculitis in glomerular capillaries.1,2 In the acute phase, the GN usually has necrosis and crescent formation and an absence or paucity of immunoglobulin deposition and is often designated pauci-immune crescentic glomerulonephritis. When occurring in the apparent absence of systemic vasculitis, pauci-immune crescentic GN is sometimes referred to as renal-limited vasculitis. Microscopic polyangiitis, GPA, EGPA, and renal-limited pauci-immune crescentic GN are all associated with ANCA.5–9 The most common antigen specificities of ANCA in patients with vasculitis and GN are for proteinase 3 (PR3) and myeloperoxidase (MPO). Recently, autoantibodies to lysosome-associated membrane protein 2 (LAMP-2) were reported in the circulation of most patients with either MPO-ANCA or PR3-ANCA.10 LAMP-2 has homology to the bacterial adhesin FimH, and thus autoantibodies to LAMP-2 may arise by molecular mimicry secondary to infection with fimbriated gram-negative bacteria. Further, rats injected with anti–LAMP-2 or immunized with FimH develop pauci-immune focal necrotizing and crescentic GN. At least one research group was not able to reproduce these results.11 These observations are intriguing but need additional confirmation. The strong association of ANCA with this distinctive form of small-vessel vasculitis suggests that ANCAs are involved in the pathogenesis.5–9 The report of a neonate who developed GN and pulmonary hemorrhage possibly caused by transplacental passage of MPO-ANCA IgG is intriguing but has not been substantiated by additional reports.12 The observation that ANCA titers correlate with disease activity also suggests a pathogenic role; however, this correlation is not strong, and some patients with clinically and pathologically typical MPA, GPA, or renal-limited pauci-immune crescentic GN are negative using conventional serologic testing for ANCA. A recent report explains both these discrepancies, at least in patients with MPO-ANCA, based on a multiplicity of ANCA epitope specificities.13 The MPO-ANCA epitope specificity determines not only the pathogenicity but also the detectability and clinical predictive value of circulating MPO-ANCA. For example, ANCAs with certain epitope specificities only occur in patients with active disease, whereas other MPO-ANCA epitope specificities occur not only in patients with active disease, but also in patients in remission and even in healthy controls (natural ANCA), although at very low titers. Some AAV patients who are negative by conventional serologic testing have MPO-ANCA with very restricted epitope specificity that can be detected with special unmasking techniques.13 The pathogenic potential of ANCA is supported by the observation that administration of certain drugs, such as propylthiouracil, hydralazine, and penicillamine, can induce AAV.14 Cocaine adulterated with levamisole also can induce AAV associated with high titers of MPO-ANCA, PR3-ANCA, and ANCA specific for another neutrophil granule protein elastase.15 Levamisole-induced vasculitis has frequent cutaneous leukocytoclastic angiitis and upper respiratory tract involvement, but rarely renal or lung involvement. A number of in vitro observations suggest mechanisms by which ANCA can cause vascular injury.6,7 Priming of neutrophils by cytokines, as would occur with a viral infection, causes neutrophils to increase expression of ANCA antigens on their surfaces, where they are accessible to interact with ANCA. Cytokine-primed neutrophils that are exposed to ANCA release IgG from granules, release toxic oxygen metabolites, and kill cultured endothelial cells.6,7,16–18 ANCA-antigen complexes adsorb onto endothelial cells, where they could participate in in situ immune complex formation.19 ANCA activation of neutrophils is mediated by both F(ab)′2 binding to neutrophils and Fc receptor engagement.20,21 Neutrophils that have been activated by ANCA adhere to endothelial cells and release mediators of inflammation and cell injury.17,18 If occurring in vivo, these events would lead to vasculitis as a result of neutrophils adhering to, penetrating, and destroying vessel walls (Fig. 25-3).22 The ability of ANCA IgG to cause pauci-immune necrotizing and crescentic GN and vasculitis has been demonstrated in multiple animal models induced with MPO-ANCA, although no widely accepted model of PR3-ANCA disease has been developed. Wild-type or immunodeficient mice that receive anti-MPO antibodies intravenously develop pauci-immune focal necrotizing GN with crescents.23A rat model of pauci-immune necrotizing and crescentic GN has been developed by immunizing rats with human MPO, resulting in the development of antibodies that cross-react with rat MPO and are able to induce pauci-immune glomerular necrosis and crescents.24 MPO-ANCA GN in mice is mediated by neutrophil activation, modulated by Fc gamma receptor repertoire, and can be prevented by neutrophil depletion.6,7,25 Activation of the alternative complement pathway plays a role in amplifying ANCA-induced inflammation.26 ANCA-activated neutrophils release factors that activate the alternative complement pathway, resulting in the generation of C5a that is strongly chemotactic for neutrophils and primes neutrophils to facilitate further activation by ANCA.27,28 The relevance of these experimental observations in patients is supported by a report that AAV patients have increased plasma levels of alternative pathway activation markers C3a, C5a, soluble C5b-9, and Bb during active disease but no remission, and that the plasma level of Bb correlated with percentage of cellular crescents in the renal biopsies and with Birmingham Vasculitis Activity Scores.29 Thus, the clinical and experimental data indicate that ANCA can activate neutrophils and cause vasculitis, especially if there are concurrent synergistic proinflammatory stimuli. The requirement for a synergistic inflammatory process may be reflected in the frequent association of the onset of ANCA small-vessel vasculitis with an influenza-like syndrome.30 An influenza-like syndrome is a manifestation of high levels of circulating cytokines that could serve as priming factors for neutrophils, making them more receptive to activation by ANCA. Wegener granulomatosis (GPA), MPA, and EGPA usually begin during the fifth, sixth, and seventh decades of life but may occur at any age. There is a slight male predominance. The incidence is disproportionately greater in Caucasians than in African Americans. In Europe, MPA has a prevalence of approximately 2.5 in 100,000; GPA, 2.5 in 100,000; and EGPA, 1 in 100,000 population.31 GPA is more common in colder than in warmer climates, whereas MPA has the opposite trend. AAV in Asia is much more often associated with MPO-ANCA than PR3-ANCA.32 Generalized nonspecific manifestations of systemic inflammatory disease, such as fever, malaise, anorexia, weight loss, myalgias, and arthralgias, often are present. Many patients with vasculitis trace the onset of their disease to a flu-like illness.30 The clinical manifestations of GPA, MPA, and EGPA are extremely varied because they are influenced by the sites of involvement and the activity versus the chronicity of involvement. All three categories of vasculitis share features caused by the small-vessel vasculitis, and patients with GPA and EGPA have the additional features that define each of these syndromes.3,4,30,33,34 Renal involvement occurs often in GPA and MPA and less frequently in EGPA (Table 25-2). The most common renal manifestations are caused by glomerular involvement and include hematuria, proteinuria, and renal failure. The renal failure often has the characteristics of rapidly progressive glomerulonephritis (RPGN) in GPA and MPA but usually is less severe in EGPA. Patients with GPA and MPA also can present with a subacute or chronic nephritis. A cohort of more than 300 pauci-immune crescentic GN patients evaluated at renal biopsy had a mean age of 56 ± 20 years (range 2 to 92 years), male-to-female ratio of 1.0 : 0.9, mean serum creatinine concentration of 6.5 ± 4.0 mg/dl (range 0.8 to 22.1 mg/dl), and proteinuria of 1.9 ± 3.0 g/day (range 0.1 to 18 g/day).35 Table 25-2 Organ system involvement in small-vessel vasculitis. GPA, Granulomatosis with polyangiitis; EGPA, eosinophilic granulomatosis with polyangiitis; HSP, Henoch-Schönlein purpura. (Modified from reference 2.) Cutaneous involvement occurs frequently in vasculitis. Purpura is a common manifestation of GPA, MPA, and EGPA (Fig. 25-4). The purpura is most common on the lower extremities and tends to occur as recurrent crops. The purpura may be accompanied by small areas of ulceration. Nodular cutaneous lesions are much more frequent in GPA and EGPA than MPA. Nodules can be caused by dermal or subcutaneous arteritis and the necrotizing granulomatous inflammation. Upper respiratory tract involvement is most common in GPA and EGPA but also occurs in MPA.36 In all three categories, patients can have pulmonary hemorrhage caused by hemorrhagic alveolar capillaritis. Patients with GPA and EGPA also can have pulmonary injury caused by necrotizing granulomatous inflammation, which may be detected radiographically as nodular or cavitating lesions. By definition, patients with MPA do not have granulomatous respiratory tract lesions.4 Manifestations of upper respiratory tract disease include subglottic stenosis, sinusitis, rhinitis, nasal septal collapse, otitis media, and ocular inflammation. These features are most common in GPA but may occur in EGPA and MPA. The upper respiratory inflammation in MPA is caused by angiitis alone, without granulomatous inflammation. Destruction of bone, for example, resulting in septal perforation and saddle nose, appears to require necrotizing granulomatous inflammation and therefore does not occur in MPA. When patients with saddle nose have ANCA, it is almost always PR3-ANCA.36 Cardiac disease is identified in approximately 50% of patients with EGPA but in less than 20% of patients with GPA or MPA. Manifestations range from transient heart block and ventricular hypokinesis that respond to immunosuppressive treatment to infarction and severe life-threatening myocarditis. Pericarditis and endocarditis also may occur. Peripheral neuropathy, usually with a mononeuritis multiplex pattern, is the most common neurologic manifestation and is most frequent in EGPA. Central nervous system involvement is less common and most often is manifested as vasculitis within the meninges. Gastrointestinal involvement typically causes abdominal pain and blood in the stool, with mesenteric ischemia and rarely intestinal perforation. Vasculitis in the pancreas and liver can mimic pancreatitis and hepatitis symptomatically and cause elevated serum pancreatic and liver enzymes. Serologic testing for ANCA is a useful diagnostic procedure for pauci-immune small-vessel vasculitis and pauci-immune crescentic GN but should be interpreted in the context of other characteristics of the patient.37–41 Laboratory testing for ANCA should include both indirect immunofluorescence microscopy assay (IFA) and enzyme immunoassay (EIA).38 IFA using normal human neutrophils as substrate produces two major staining patterns (Fig. 25-5): cytoplasmic (c-ANCA), in which staining occurs diffusely throughout the cytoplasm, and perinuclear (p-ANCA). By EIA, most c-ANCA have specificity for protease 3 (PR3-ANCA), and most p-ANCA have specificity for myeloperoxidase (MPO-ANCA). For adequate diagnostic accuracy, all serologic testing for ANCA should include an immunochemical analysis for antigen specificity, such as an EIA.38 Although positive results are rare in completely healthy individuals, approximately one fourth of patients with other inflammatory renal diseases (especially lupus) will have a false-positive IFA result (usually with a p-ANCA pattern), and approximately 5% will have a false-positive EIA result (usually at low titer).39 ANCA testing has good sensitivity for AAV (80% to 90%). The specificity and predictive value depend on the population of patients and the quality of the assay.37,41 Although ANCAs are most frequent in patients with pauci-immune crescentic GN, one fourth to one third of patients with anti-GBM crescentic GN and one fourth of those with idiopathic immune complex crescentic GN with 50% or more crescents are ANCA positive.35,42 Some of these patients have well-recognized types of immune complex GN complicated by ANCA, such as membranous glomerulopathy and IgA nephropathy, whereas others have nonlupus IgG-dominant immune complex disease that cannot be categorized further. Patients with concurrent ANCA and anti–glomerular basement membrane (anti-GBM) antibodies have a worse prognosis than patients with ANCA alone. Table 25-3 provides an estimate of the relative frequencies of PR3-ANCA/c-ANCA and MPO-ANCA/p-ANCA in the different clinical phenotypes of AAV. PR3-ANCA/c-ANCA are most prevalent in GPA, and MPO-ANCA/p-ANCA are most prevalent in renal-limited pauci-immune crescentic GN and EGPA. Patients with MPA have a more even distribution of PR3-ANCA/c-ANCA and MPO-ANCA/p-ANCA, although this varies among geographic regions as stated earlier. Patients with EGPA have the lowest overall frequency of ANCA, but the frequency of ANCA is much higher in patients with GN (75%) than in those with no GN (26%).43 The ANCA specificity correlates with clinical symptoms, with PR3-ANCA having the highest frequency (~90% when ANCA present) in patients who have destructive upper respiratory disease, especially saddle nose.36 Changes in ANCA titers over time correlate to a degree with disease activity but are not infallible markers and thus must be interpreted with caution.39,44 In general, titers decrease with treatment and increase before or at disease recurrence. An increase in ANCA titer should prompt careful evaluation of the patient for corroborating evidence of exacerbation, but most physicians do not modify treatment on the basis of an increase in titer without accompanying clinical signs. A recent report suggests that epitope-specific assays may provide much better correlation with and prediction of disease outcome.13 MPO-ANCAs with certain epitope specificities occur only in patients with active disease, disappear with remission, and reappear during relapse, whereas MPO-ANCAs with other specificities remain during disease remission. From 10% to 20% of patients with pauci-immune necrotizing and crescentic GN and pauci-immune small-vessel vasculitis will be ANCA negative. The clinicopathologic and outcome characteristics of these patients are no different from those of ANCA-positive patients.45 In the same epitope-specific assays previously mentioned, some AAV patients who are negative by current clinical assays have MPO-ANCA with restricted epitope specificity that can be detected with sensitive assays that remove a masking factor.13 ANCA may be positive in inflammatory conditions other than vasculitis, including inflammatory bowel disease (IBD), rheumatoid disease, chronic inflammatory liver disease, bacterial endocarditis, and cystic fibrosis. In IBD, specificity of the ANCA may not be against PR3 or MPO but against other neutrophil antigens, including lactoferrin, cathepsin G, and anti–bactericidal/permeability-increasing protein (BPI). The acute vascular lesion of the pauci-immune small-vessel vasculitides is segmental fibrinoid necrosis, often accompanied by leukocyte infiltration and leukocytoclasia1,2,46–49 (leukocyte fragmentation; Figs. 25-6 and 25-7). The earliest vasculitic lesions have infiltrating neutrophils that are quickly replaced by predominantly mononuclear leukocytes. The acute necrotizing lesions evolve into sclerotic lesions and may be complicated by thrombosis.
Renal and Systemic Vasculitis
Definition
Vasculitis Categories and Definitions
Category/Name
Definition
Large-Vessel Vasculitis
Takayasu arteritis
Arteritis, often granulomatous, predominantly affecting aorta and/or its major branches. Onset usually in patients younger than 50.
Giant cell arteritis
Arteritis, often granulomatous, usually affecting aorta and/or its major branches, with predilection for branches of carotid and vertebral arteries; often involves temporal artery. Onset usually in patients older than 50 and often associated with polymyalgia rheumatica.
Medium-Vessel Vasculitis
Polyarteritis nodosa
Necrotizing arteritis of medium or small arteries without glomerulonephritis (GN) or vasculitis in arterioles, capillaries, or venules; and not associated with ANCA.
Kawasaki disease
Arteritis associated with mucocutaneous lymph node syndrome and predominantly affecting medium and small arteries; coronary arteries are often involved; aorta and large arteries may be involved. Usually occurs in infants and young children.
Small-Vessel Vasculitis
ANCA-Associated Small-Vessel Vasculitis
Necrotizing vasculitis, with few or no immune deposits, predominantly affecting small vessels (capillaries, venules, arterioles, small arteries), associated with MPO-ANCA or PR3-ANCA. Not all patients have ANCA. Add prefix indicating ANCA reactivity, e.g., PR3-ANCA, MPO-ANCA, ANCA-negative.
Microscopic polyangiitis (MPA)
Necrotizing vasculitis, with few or no immune deposits, predominantly affecting small vessels (capillaries, venules, arterioles). Necrotizing arteritis involving small and medium arteries may be present. Necrotizing GN is common. Pulmonary capillaritis often occurs. Granulomatous inflammation is absent.
Granulomatosis with polyangiitis (Wegener) (GPA)
Necrotizing granulomatous inflammation usually involving upper and lower respiratory tract, and necrotizing vasculitis affecting predominantly small to medium vessels (capillaries, venules, arterioles, arteries, veins). Necrotizing GN is common.
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA)
Eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium vessels, and associated with asthma and eosinophilia. ANCA is more common when GN is present.
Immune Complex Small-Vessel Vasculitis
Vasculitis with moderate to marked vessel wall deposits of immunoglobulin and/or complement components predominantly affecting small vessels (capillaries, venules, arterioles, small arteries). GN is common.
Anti–glomerular basement membrane (anti-GBM) disease
Vasculitis affecting glomerular capillaries, pulmonary capillaries, or both, with basement membrane deposition of anti–basement membrane autoantibodies. Lung involvement causes pulmonary hemorrhage, and renal involvement causes GN with necrosis and crescents.
Cryoglobulinemic vasculitis
Vasculitis with cryoglobulin immune deposits affecting small vessels (predominantly capillaries, venules, or arterioles) and associated with cryoglobulins in serum. Skin, glomeruli, and peripheral nerves are often involved.
IgA vasculitis (IgAV) (Henoch-Schönlein purpura)
Vasculitis, with IgA1-dominant immune deposits, affecting small vessels (predominantly capillaries, venules, or arterioles). Often involves skin and gastrointestinal tract and frequently causes arthritis. GN indistinguishable from IgA nephropathy may occur.
Hypocomplementemic urticarial vasculitis (anti-C1q vasculitis)
Vasculitis accompanied by urticaria and hypocomplementemia affecting small vessels (capillaries, venules, arterioles), and associated with anti-C1q antibodies. GN, arthritis, obstructive pulmonary disease, and ocular inflammation are common.
Small-Vessel Vasculitis
Medium-Vessel Vasculitis
Large-Vessel Vasculitis
Small-Vessel Pauci-Immune Vasculitis
Pathogenesis
Epidemiology
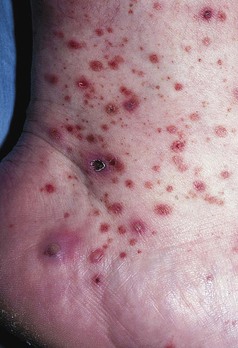
Clinical Manifestations
Organ System Involvement in Small-Vessel Vasculitis
Organ System
Frequency of Involvement (%)
Microscopic Polyangiitis
GPA (Wegener)
EGPA (Churg-Strauss)
IgA Vasculitis (HSP)
Cryoglobulinemic Vasculitis
Kidney
90
80
45
50
55
Skin/cutaneous
40
40
50
90
90
Lungs
50
90
90
<5
<5
Ear, nose, throat
35
90
50
<5
<5
Musculoskeletal
60
60
50
75
70
Neurologic
30
50
60
10
40
Gastrointestinal
50
50
70
60
30
Antineutrophil Cytoplasmic Autoantibody
Pathology