Matthew J. Cervelli, Graeme R. Russ
Principles of Drug Therapy, Dosing, and Prescribing in Chronic Kidney Disease and Renal Replacement Therapy
Renal impairment can alter drug pharmacokinetics and pharmacodynamics, and consequently patients with renal impairment are at risk of adverse effects. In addition, these patients take multiple drugs and are at high risk of drug interactions and drug-related problems.1 To prescribe safely and effectively, clinicians should be familiar with the pharmacokinetics of drugs in varying stages of renal impairment and renal replacement therapy (RRT) and ideally rely on data from these populations. Unfortunately, such information is not always available, and exclusion of these patients from clinical studies can lead to restrictive licensed recommendations. This chapter describes pharmacokinetic principles and highlights common prescribing issues in patients with renal impairment, dialysis, and transplantation. Specific dose recommendations and pharmacokinetic data are not included but can be obtained from references developed specifically to provide concise, reliable, and practical information for prescribing the settings of renal impairment, dialysis, and transplantation.2–5
Pharmacokinetic Principles
Pharmacokinetics describes the behavior of a drug (or metabolite) with regard to absorption, distribution, metabolism, and elimination (Fig. 77-1 and Table 77-1).6,7
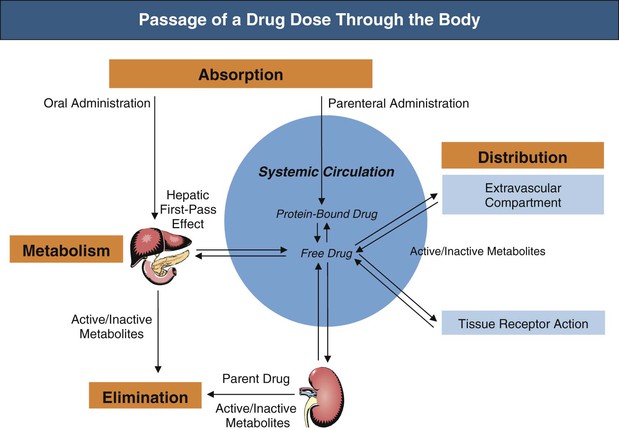
Table 77-1
Pharmacokinetic parameters.
| Pharmacokinetic Parameters | ||
| Parameter | Definition | Application |
| Bioavailability (F) | Percentage of a dose that appears in the systemic circulation after administration by a nonintravenous route | Determines the amount of drug reaching the systemic circulation |
| Volume of distribution (VD) | Proportionality constant relating the amount of drug in the body at a given time to a simultaneously occurring drug concentration in plasma, blood, or other reference fluid at an identical time | Determines the size of loading doses |
| Clearance (Cl) | Proportionality constant between rate of drug elimination from the body (units = mass/unit time) and the concentration of the drug in plasma or blood at the same point in time | Determines the maintenance dose |
| Half-life (t1/2) | Time taken for the drug concentration in plasma to fall to half its current value | Determines frequency of administration and time to steady state |
Absorption: Bioavailability
Bioavailability (F) is the portion of a drug dose that appears in the systemic circulation after administration by a nonintravenous route. Whereas drugs given intravenously have 100% bioavailability, drugs given by alternative routes pass through a series of biologic membranes before entering the systemic circulation so that only a fraction may reach the circulation.
After oral administration, the liver can metabolize a drug during “first pass” when it is absorbed or later when it is delivered through systemic blood flow. First-pass metabolism can significantly reduce absorption. The gastrointestinal mucosa also acts as a barrier to absorption by metabolizing drugs or retarding absorption.8 Renal impairment can influence absorption, although the effect is difficult to quantify and clinical examples are limited. Gastrointestinal edema can limit oral absorption, for example, of furosemide. Nausea and vomiting from uremia can impair absorption and contact time between the drug and gastrointestinal mucosa. In patients with advanced uremia, the alkalinizing effect of salivary urea may decrease absorption of drugs optimally absorbed in an acid milieu. Commonly prescribed metallic ion phosphate binders can decrease drug absorption by forming nonabsorbable complexes with drugs (Table 77-2).9 Changes in cardiac output in renal failure can reduce the rate and extent of absorption for drugs with significant first-pass metabolism. Increased absorption in patients with renal impairment from reduced first-pass metabolism is seen with some β-blockers, dextropropoxyphene, and dihydrocodeine. Comorbidities in renal patients also have an effect; for example, absorption can be erratic because of diabetic gastrointestinal neuropathy.
Table 77-2
Effect of food and phosphate binders on oral drug absorption.
| Effect of Food and Phosphate Binders on Oral Drug Absorption | |
| Drug | Effect of Food |
| Captopril | Decreases serum drug levels |
| Bisphosphonates (oral) | Significantly reduces drug absorption |
| Cinacalcet | Significantly increases drug absorption |
| Iron (oral) | Decreases absorption |
| Ketoconazole or itraconazole | Increases absorption with reduced pH |
| Sirolimus | High-fat meals increase absorption |
| Tacrolimus | Reduces drug absorption |
| Drug | Effect of Metallic Phosphate Binders |
| Bisphosphonates (oral) | Calcium-containing binders significantly reduce absorption |
| Fluoroquinolones | Reduction in absorption |
| Tetracycline | Reduction in absorption |
| Thyroid hormones | Reduction in absorption |
Distribution
Volume of Distribution
After absorption, drugs may distribute from plasma to an extravascular compartment. Each drug has a characteristic volume of distribution (VD), which is really an apparent VD because it does not correspond to an anatomic space but instead relates the amount of drug in the body to its plasma concentration. VD is used to calculate the loading dose to achieve a desired plasma concentration (VD = dose/plasma). Water-soluble drugs tend to be restricted to the extracellular fluid space and have a relatively small VD. Lipid-soluble drugs penetrate body tissues and have a large VD. Increased VD can occur with edema, ascites, or infection, particularly for water-soluble drugs. If usual doses are given, low concentrations result. Conversely, muscle wasting or volume depletion can decrease the VD of water-soluble drugs, and usual doses produce high concentrations.
Plasma Protein Binding
Drugs can bind extensively to plasma proteins.10 The free (unbound) fraction of a drug is usually the portion that exerts a pharmacologic effect. If protein binding is reduced, a greater free fraction is available for any given total drug concentration, which may increase drug activity. Organic acids usually have a single binding site on albumin, whereas organic bases have multiple binding sites on glycoproteins. Protein binding can be altered in patients with renal impairment, especially when serum albumin is low (e.g., nephrotic syndrome) or when uremic toxins displace drugs from binding sites (Table 77-3). Predicting the effect of changes in protein binding is difficult because even though more free drug is available at the site of action, more is available for metabolism or renal excretion. Hence, lower plasma concentrations can occur and drug half-life may decrease rather than increase. Phenytoin, for example, has marked decreases in protein binding in patients with renal impairment, and toxicity can occur despite normal or low total plasma concentrations because of an increase in free fraction. With albuminuria, bound drug may also be lost, which may partially explain the refractoriness of nephrotic patients to diuretics. In patients with chronic kidney disease (CKD), high plasma levels of α1-acid glycoprotein are induced in acute and chronic inflammation, which can increase drug binding.
Table 77-3
Protein binding of drugs in renal disease.
↓ indicates reduced protein binding in renal impairment. For all other drugs listed, protein binding is increased. The therapeutic effect is, however, not easily predicted (see text).
| Protein Binding of Drugs in Renal Disease | |
| Major Effects | Minor Effects |
| Albumin: Binding Sites for Acidic Compounds | |
| Barbiturates (↓) | Ascorbic acid |
| Benzodiazepine | Valproate (↓) |
| Carbamazepine | Fatty acids |
| Fibrates | Nafcillin |
| Furosemide (↓) | Phenylbutazone (↓) |
| Mycophenolate mofetil | Probenecid |
| Penicillins | Thiopental (↓) |
| Phenytoin (↓) | Warfarin (↓) |
| Sulfonamides (↓) | Thyroxine (↓) |
| Globulins: Binding Site for Basic Compounds | |
| Digoxin (↓) | Adenosine |
| Methadone | Amitriptyline |
| Propranolol | Chloramphenicol |
Metabolism
Drug metabolism is primarily a hepatic function by which drugs are converted to more water-soluble entities to promote elimination by the kidneys and bile. Despite the assumption that nonrenal clearance is unchanged, renal impairment can alter and slow drug metabolism.11 It is important to note that some drugs have renally cleared active or toxic metabolites that, although insignificant in normal renal function, can accumulate in patients with renal impairment.
Elimination
The kidney is the most important organ for drug and metabolite elimination. Terms to describe drug clearance are shown in Box 77-1. Total drug clearance equals the apparent volume of blood or plasma from which the drug is cleared per unit of time and is expressed as the dose divided by the area under the drug concentration curve (AUC). Half-life describes the time taken for plasma concentrations to halve and is related to VD and clearance. Quantitation of drug elimination by the kidney is expressed as renal clearance, which depends on renal blood flow and the ability of the kidney to remove the drug. Renal drug clearance is the balance of its glomerular filtration rate (GFR), renal tubular secretion, and tubular reabsorption. Glomerular filtration depends on molecular size (<10 kd), charge, and protein binding (increased when binding decreases). Secretion of drugs eliminated by tubular transport may change with renal disease, but measurement of tubular function is difficult. Practically, as GFR decreases, drugs dependent on tubular secretion are also excreted more slowly. Assuming no change in nonrenal clearance, as GFR falls, clearance of drugs (and metabolites) eliminated by the kidney decreases, and their half-life is prolonged.
Box 77-1 Mathematics of drug elimination.
AUC, Area under the concentration-time curve; VD, volume of distribution (dose/blood concentration).
Mathematics of Drug Elimination
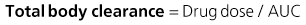
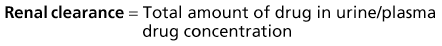
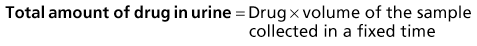
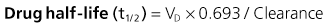
Prescribing Principles for Chronic Kidney Disease and Renal Replacement Therapy
Ideally, dose modification recommendations should be made by comparing the pharmacokinetics of a drug at varying stages of renal impairment relative to normal function. However, lack of reliable data and individual patient factors limit such generalities, and clinical judgment about a patient’s ability to handle a drug is vital.12 Dose nomograms, tables, and computer recommendations are helpful but not necessarily associated with better outcomes. Physicians should use clinical judgment to evaluate every situation individually, choose a dosage regimen based on factors in that patient, and continually reevaluate response to therapy.
Initial Assessment and Laboratory Data
A targeted history is important in assessing dose in patients with renal impairment. Previous drug efficacy or toxicity should be determined and the current drug list reviewed for potential interactions or nephrotoxins. Physical and laboratory parameters indicate volume status, height, weight, and extrarenal disease (e.g., liver).
Estimating Renal Function for Drug Dosage
Estimating renal function is essential in renal drug dosage. The greater the degree of renal impairment, the greater the potential for dose modification. With exceptions, dose modification is usually not clinically necessary until GFR is below 30 ml/min. Assessment of renal function for drug administration is not a precise science, and what is essential for clinical decision making is awareness that renal function is impaired, and approximately to what extent, rather than knowledge of the precise GFR.13 For drug prescribing, renal impairment is usually graded as mild (30 to 60 ml/min), moderate (10 to 30 ml/min), or severe (dialysis). For drug dosage calculation, methods of estimating GFR are sufficient (see Chapter 3). The Cockcroft-Gault equation has been the most widely used and accepted method for drug dosage calculation. The Modification of Diet in Renal Disease (MDRD) formula, if not corrected for body surface area, can lead to recommendations different from those obtained by the Cockcroft-Gault equation. An important limitation of many renal function estimates is inaccuracy of single-point estimates when renal function is rapidly changing. This may lead to overestimation or underestimation of renal function and underdose or overdose. In patients with severe acute kidney injury (AKI), the decline in GFR is so rapid that patients should receive dosages appropriate for a GFR below 10 ml/min. The opposite is true in rapidly improving renal function after AKI or early post-transplant.
Activity and Toxicity of Metabolites
It is essential to consider the activity (or toxicity) of drug metabolites in addition to that of the parent drug itself. Renally cleared metabolites can accumulate, leading to enhanced drug action or toxicity (Table 77-4).
Table 77-4
Drugs with renally cleared active or toxic metabolites.
CNS, Central nervous system.
| Drugs with Active or Toxic Metabolites That Accumulate in Subjects with Reduced GFR | ||
| Drug | Active Metabolite | Consequence |
| Allopurinol | Oxypurinol | |
| Cefotaxime | Desacetylcefotaxime | |
| Glyburide | 4-trans-Hydroxyglibenclamide 3-cis-Hydroxyglibenclamide | Hypoglycemia |
| Morphine | Morphine-6-glucuronide | CNS side effects |
| Tramadol | O-Desmethyltramadol | CNS side effects |
| Venlafaxine | O-Desmethylvenlafaxine | CNS and cardiovascular side effects |
| Drug | Toxic Metabolite | Consequence |
| Dapsone | Monoacetylated metabolite | |
| Meperidine (pethidine) | Normeperidine (norpethidine) | CNS (seizures) |
| Nitroprusside | Thiocyanate | Cyanide toxicity |
| Procainamide | N-Acetylprocainamide (NAPA) | Arrhythmia |
| Propoxyphene | Norpropoxyphene | Cardiac toxicity |
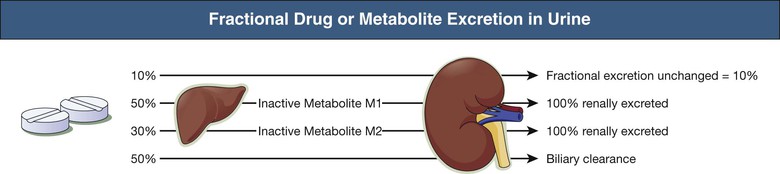
Fraction of Active Drug (and Active or Toxic Metabolite) Excreted Unchanged in Urine
The greater the fraction of active drug or metabolite excreted unchanged by the kidneys (fe), the greater the need for dose modification. It is usually clinically necessary to modify doses only if the fe is greater than 25% to 50%. The reported fe is often determined from studies that do not distinguish between parent drug and metabolites. The contribution of inactive nontoxic metabolites to overall renal drug elimination may exaggerate the potential for harm. Active or toxic metabolites should be assessed separately for their dependence on renal elimination in the same way as the parent drug (Fig. 77-2).
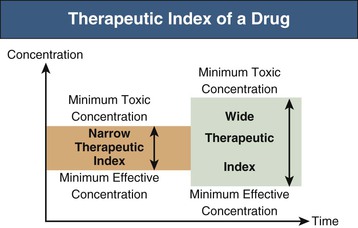
Therapeutic Index of the Drug or Metabolites
The decision to modify dosage for patients with renal impairment is influenced by the therapeutic window or index of the drug. The therapeutic window is the range of plasma drug concentrations spanning the minimum concentration for clinical efficacy and toxicity. The therapeutic index is the ratio of these concentrations (Fig. 77-3). If the therapeutic window is wide (e.g., many penicillins), there may be no clinical need for dose modification despite significant renal elimination. If the therapeutic window is narrow (e.g., digoxin), dose modification is more critical. Clinicians should judge the clinical relevance of increased exposure to drug or metabolites.
Avoid Nephrotoxic Drugs
A wide range of drugs can cause nephrotoxicity (Table 77-5). Idiosyncratic nephrotoxicity (e.g., interstitial nephritis) is unpredictable and independent of dose. Predictable hemodynamic-related nephrotoxicity can occur with angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), nonsteroidal anti-inflammatory drugs (NSAIDs), diuretics, antihypertensives, and laxatives. Direct tubular nephrotoxins include aminoglycosides, vancomycin, amphotericin, cisplatin, calcineurin inhibitors (CNIs), and radiographic contrast media. Obstructive uropathy can occur with tubular crystallization of acyclovir, statin-induced rhabdomyolysis, or tricyclic antidepressant use. In dialysis patients with no significant residual renal function, use of nephrotoxic drugs may be acceptable. Drug nephrotoxicity is discussed further in Chapter 69.
Table 77-5
Some examples of nephrotoxic drugs.
ACE, Angiotensin-converting enzyme; AKI, acute kidney injury; ARBs, angiotensin receptor blockers; GFR, glomerular filtration rate; 20-HETE acid, 20-hydroxyeicosatetraenoic acid; H2O2; hydrogen peroxide; mTOR, mammalian target of rapamycin; NSAIDs, nonsteroidal anti-inflammatory drugs; PG, prostaglandin.
| Nephrotoxic Drugs | ||
| Examples | Mechanism | Prevention and Management |
| ACE inhibitors, ARBs | Impairment of angiotensin II–mediated afferent arteriole dilation during renal hypoperfusion | Withdraw in renal hypoperfusion |
| Aminoglycosides Amikacin Gentamicin Tobramycin | In proximal tubules, aminoglycosides bind to anionic phospholipid, are delivered to megalin, are taken up into the cell, accumulate, and cause direct toxicity | Alternative if possible Monitor drug concentrations Avoid multiple daily doses Withdraw if creatinine rises |
| Antifungals Amphotericin | Afferent vasoconstriction and direct action to reduce GFR Distal tubular injury via creation of pores that increase membrane permeability leading to hypokalemia, hypomagnesemia, metabolic acidosis caused by tubular acidosis, polyuria from nephrogenic diabetes insipidus | Avoid use Administer slowly with hydration Use liposomal preparation |
| Antivirals | ||
| Acyclovir | Deposition of drug crystals → intratubular obstruction and foci of interstitial inflammation | Avoid bolus dose Reduce dose in renal impairment Hydrate during therapy |
| Cidofovir | Induces apoptosis in proximal tubule → tubular dysfunction, diabetes insipidus, renal failure | Oral probenecid and hydration |
| Foscarnet | Direct tubular toxicity → acute tubular necrosis, nephrogenic diabetes insipidus Crystals in glomerular capillary lumen and proximal tubular lumen | Hydration |
| Indinavir | Crystal neuropathy, nephrolithiasis → obstructive AKI | Hydration |
| Calcineurin inhibitors Cyclosporine Tacrolimus | ↓ PG and ↑ 20-HETE acid production → vasoconstriction, generation of H2O2 resulting in depleted glutathione → decreased GFR, ischemic collapse or scarring of the glomeruli, vacuolization of the tubules, and focal areas of tubular atrophy and interstitial fibrosis | Measure plasma concentrations Avoid interacting drugs Withdraw drug (switch to mTOR inhibitor) |
| Chemotherapeutics | ||
| Cisplatin | Cis chloride replaced by H2O → highly reactive OH radical → DNA injury, tubular cell death Nephrogenic diabetes insipidus, hypomagnesemia (may be persistent) | Forced diuresis and hydration |
| Ifosfamide | Direct tubular injury and mitochondrial damage → renal tubular acidosis, Fanconi-like syndrome, nephrogenic diabetes insipidus hypokalemia | |
| Intravenous immunoglobulin (sucrose-containing products) | Accumulation of sucrose in proximal convoluted tubules, ↑ osmolarity → cell swelling, vacuolization, and tubular luminal occlusion | Infusion rate <3 mg sucrose/kg/min Avoid radiocontrast Avoid sucrose-containing product Hydration |
| Lithium | Impairment of collecting duct concentrating ability → diabetes insipidus Chronic tubulointerstitial nephropathy (tubular atrophy and interstitial fibrosis) | Measure plasma concentrations Prevent dehydration Avoid thiazides |
| NSAIDs | Hemodynamically induced AKI caused by vasoconstriction via reduced prostaglandin production | Avoid use Withdraw during hypoperfusion |
| Recruitment and activation of lymphocytes → acute and chronic tubulointerstitial nephritis, with or without nephrotic syndrome | Withdraw (add corticosteroids) | |
| Proton pump inhibitors | Interstitial nephritis | Withdraw (add corticosteroids) |
| Radiocontrast media | High osmolarity, medullary vasoconstriction, ↑ active transport in thick ascending loop of Henle → ↑ O2 demand | Hydration preprocedure and postprocedure Acetylcysteine |
| Sulfonamides | Intrarenal precipitation → Kidney stone formation | Fluid intake >3 l/day; monitor urine for crystals Alkalinize urine to >7.15 if crystal seen |
Drugs That Aggravate the Metabolic Effects of Renal Impairment
Some drugs have no direct adverse effect on renal function but when used in patients with renal impairment can aggravate the metabolic consequences of renal failure. Hyperkalemia is worsened with potassium supplements, potassium-sparing diuretics, aldosterone antagonists, and blockers of the renin-angiotensin system. The catabolic effects of tetracycline can exacerbate uremia. Sodium-containing drugs and those that promote sodium and water retention should be used cautiously because they may provoke fluid overload and hypertension.
Effect of Renal Impairment on Pharmacodynamic or Physiologic Mechanisms
Renal disease may alter a pharmacodynamic response or physiologic process, which in turn affects clinical response. For example, the inability of impaired kidneys to activate vitamin D precursors means that vitamins D2 and D3 may be less effective. Patients with renal impairment often have a coagulopathy from the effects of uremia on platelet function and may be more prone to the bleeding complications of anticoagulant and antiplatelet therapy.
Effect of Renal Impairment on the Concentration of Drug at the Site of Action
Renal impairment can alter drug concentration at the site of action. Some diuretics and antibiotics become ineffective in patients with renal impairment because they do not achieve adequate concentrations at their site of action in the renal tubules or bladder. This may preclude the use of drugs such as thiazide diuretics and nitrofurantoin or may require increased doses of others (e.g., loop diuretics).
Location of Drug Action
Drugs that have negligible bioavailability and that are used for a local or topical effect may be given safely at normal dose despite toxicity with systemic doses. These include topical NSAIDs, nebulized gentamicin, and oral vancomycin.
Method of Administration
In fluid-restricted patients, administration of intravenous drug infusions with approved fluid volumes may be undesirable. When administration exceeds daily fluid restrictions, consider alternatives or more concentrated solutions if physiochemical parameters allow. Similarly, oral drug administration with large fluid volumes (e.g., bisphosphonates) may not be advisable. In patients with severe nausea and vomiting, essential immunosuppressants should be administered intravenously.
Drug Interactions
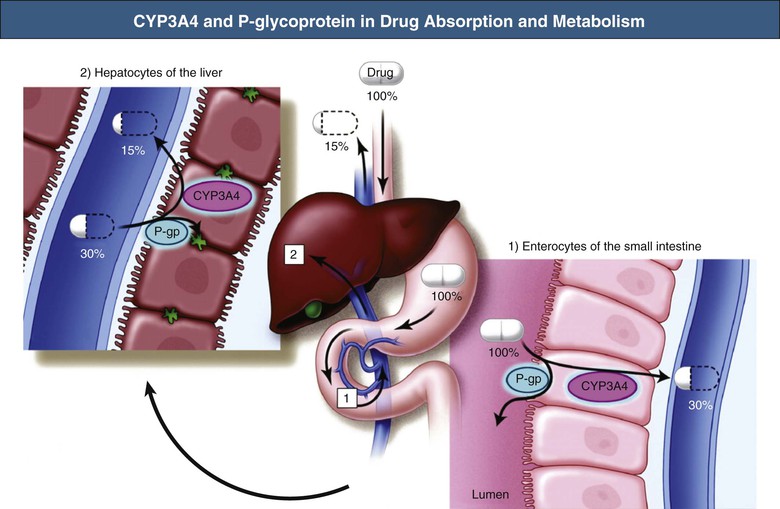
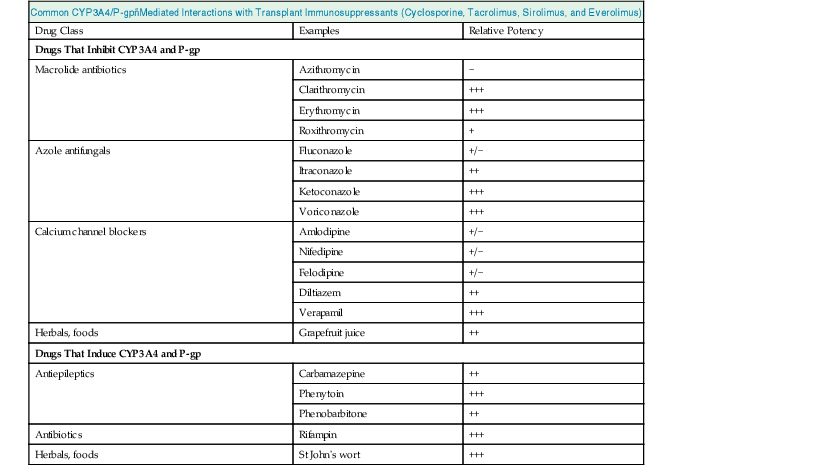
Pharmacokinetic drug interactions are frequently problematic, and awareness of clinically significant interactions is essential, especially with regard to patients receiving transplant immunosuppressants. The most important of these are cyclosporine, tacrolimus, everolimus, and sirolimus, which are substrates of both the CYP3A4 enzyme system and P-glycoprotein expressed in gastrointestinal mucosa and liver.14 Co-prescription of drugs that inhibit these systems (e.g., some azole antifungals, calcium channel blockers (CCBs), macrolides, and grapefruit juice) can increase absorption and reduce metabolism of the immunosuppressant, causing toxicity. Conversely, drugs that induce these systems (e.g., barbiturates, phenytoin, carbamazepine, rifampin, and St John’s wort [Hypericum]) can reduce absorption and increase metabolism and therefore increase the risk of rejection (Fig. 77-4 and Table 77-6). All drug changes in patients receiving transplant immunosuppression should be considered for their potential to interact, and appropriate dose modifications or alternatives used.
Clinical Condition of the Patient
The patient’s welfare should override theoretical concerns. Higher-than-recommended doses may be appropriate when there is a strong clinical indication. For example, excessive reduction in initial antibiotic doses based on renal function may be inappropriate in patients with life-threatening infection when the consequences of failed therapy are greater than potential toxicity.
Methods of Dose Reduction
Loading Doses
For most drugs, steady-state concentrations are achieved after five drug half-lives. Hence, for some drugs, a loading dose is given to reduce time to steady state. Because renal impairment may prolong the half-life, simply reducing drug doses could be a therapeutic error because this would further delay achievement of steady state. The loading dose (mg/kg) is equal to the product of the desired plasma concentration (mg/ml) and VD (ml/kg) and is independent of clearance. Provided the desired concentration and VD are unchanged, loading doses do not require modification in patients with renal impairment. In some instances, VD is altered, especially with hypoproteinemia or fluid overload, and hence some clinicians alter the loading dose of drugs with a narrow therapeutic index such as digoxin. Aminoglycoside doses may need to be increased in patients with fluid overload or sepsis who have an increased VD.
Maintenance Doses
When specific pharmacokinetic information is not available, and assuming no change in nonrenal clearance, maintenance doses should be reduced in proportion to the extent of renal impairment and renal drug elimination. For example, if renal function is 50% of normal and the drug is 100% renally excreted, a maintenance dose of 50% is required. If the drug is 50% renally cleared and the patient has 20% renal function, the dose should be 60% of normal. The dose reduction factor is estimated from first principles or the following formula:
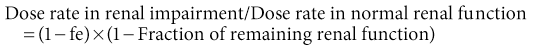
where fe is the fraction of active drug excreted unchanged in urine.
Once a dose reduction factor has been determined, the clinician must decide on a dose reduction method. Two methods are used, either alone or in combination (Fig. 77-5).
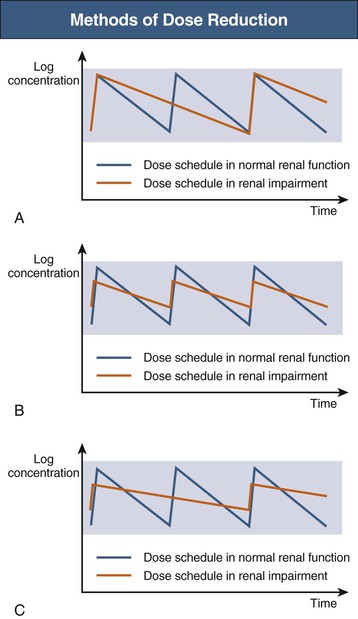
Interval Method
Because drug clearance is reduced, a reduction in delivered dose is achieved by administering the same dose less frequently. This method is particularly useful when the size of the dose and peak blood concentrations are important for efficacy (e.g., aminoglycosides). If this method is used, practical dose intervals should be recommended rather than inconvenient or complex intervals.
Dose Method
An alternative method is to administer a smaller dose at the usual interval. This method is common, especially when the size of a dose and peak concentrations are less critical for efficacy. If this method is used, clinicians should consider the availability of smaller dose formulations and the ability of the patient to accurately and safely divide available dosage forms.
Combination Method
Sometimes, especially for drugs with a narrow therapeutic index, for which tight control of concentrations is required (e.g., digoxin), a combination of the dose and interval methods is used.
Ongoing Assessment
Even with appropriate dose modification in patients with renal impairment, clinicians should always remain vigilant and closely monitor the response to therapy to guide dose titration.
Therapeutic Drug Monitoring
Therapeutic drug monitoring can provide objective information to guide dosage calculation and is valuable for drugs such as aminoglycosides, glycopeptides, digoxin, lithium, antiepileptics, and immunosuppressants (Table 77-7). Assays usually measure total blood concentrations and may significantly underestimate plasma levels of the active or free form of the drug.
Table 77-7
Therapeutic drug monitoring in renal impairment.
Target levels are dependent on assay methodology and clinical context. IV, Intravenous.
| Therapeutic Drug Monitoring in Renal Impairment | |
| Drug | Therapeutic Range and When to Draw Sample |
| Aminoglycosides (24-h dosing) | Peak (30 min after a 30-min infusion) Trough (6-12 h after dose) |
| Gentamicin | Peak: >10 mg/l |
| Tobramycin | Trough: depends on time after dose 0.5-2 mg/l |
| Amikacin | Peak: >30 mg/l Trough: depends on time after dose 1.5-6 mg/l |
| Immunosuppressants | |
| Cyclosporine | C0 (trough): 150-250 µg/ml C2 (2 h after dose): 1200-1500 µg/ml AUC0-4 >4400 µg/ml/h |
| Tacrolimus | C0 (trough): 4-12 µg/ml |
| Sirolimus | C0 (trough): 5-15 µg/ml |
| Antiarrhythmics | |
| Digoxin | 0.8-2.0 µg/ml (trough at least 6 h after dose) |
| Lidocaine | 1-5 µg/ml 8 h after IV infusion starts or is changed |
| Antipsychotics | |
| Lithium | Acute: 0.8-1.2 mmol/l (trough) Chronic: 0.6-0.8 mmol/l (trough) |
| Antiepileptics | |
| Carbamazepine | 4-12 µg/ml (trough before administration) |
| Phenytoin | 10-20 µg/ml (trough before administration) |
| Free phenytoin | 1-2 µg/ml (trough before administration) |
| Phenobarbital | 15-40 µg/ml (trough before administration) |
| Valproic acid | 40-100 µg/ml (trough before administration) |
| Vancomycin | Trough: 10-20 mg/l |
Clinical Response
Ultimately, clinical response should influence the need to modify doses. Doses should be carefully titrated according to response and adverse effects. For example, blood pressure for antihypertensives, blood glucose concentration and hemoglobin A1c (HbA1c) for oral hypoglycemics, and renal function for toxicity from CNIs, ACE inhibitors, and ARBs.
Extracorporeal Drug Losses
Failure to consider dialysis drug clearance may significantly reduce drug efficacy.5,7 Alternatively, dialysis may be used in overdose to assist drug removal (see Chapter 98). Studies of drug clearance with RRT modalities have often used variations in dialysis technique that make it difficult to compare results. Many studies before the 1990s report data from standard hemodialysis (HD) with low-flux membranes, which are less efficient at drug removal than the high-flux membranes now widely used. Many recommendations report the need for supplemental doses. Clinically, supplemental doses are rarely used, especially if less than 30% of the drug is cleared or the drug has a wide therapeutic index. Rather, drugs known to be significantly cleared by dialysis should be administered after dialysis (Table 77-8). When drugs are given in multiple daily doses, at least one of the daily doses should be administered soon after the completion of dialysis.
Table 77-8
Dialysis clearance of drugs.
LMWHs, Low-molecular-weight heparins; NSAIDs, nonsteroidal anti-inflammatory drugs; SSRIs, selective serotonin reuptake inhibitors.
| Dialysis Clearance of Commonly Prescribed Drugs | |
| Drugs Significantly Cleared by Hemodialysis That Require Administration After Dialysis or a Supplement After Dialysis | Drugs Not Significantly Cleared by hemodialysis and That Do Not Require Administration After Dialysis or a Supplement After Dialysis or That Must Be Administered at Specific Times Independent of Hemodialysis |
Antivirals | Antibiotics Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|





