Pathophysiology and Nephron Adaptation in Chronic Kidney Disease
Radko Komers
Timothy W. Meyer
Sharon Anderson
That we are endowed with a surfeit of nephrons above that needed to maintain normal homeostasis is shown by the ability of a single kidney to carry out the functions previously performed by two, after a uninephrectomy. The surplus is even more apparent in progressive chronic kidney disease (CKD), where solute and water balance are well maintained until late in the course. This chapter first reviews the compensatory changes in function and structure that occur when nephron number is reduced and that enable maintenance of homeostasis. Alterations in handling of individual solutes and water are then described. We then describe the mechanisms that underlie adaptation, and then the transition stage in which these compensatory changes can prove maladaptive and accelerate remaining nephron loss.
PRINCIPLES AND PATTERNS OF NEPHRON ADAPTATION: INTACT NEPHRONS AND GLOMERULOTUBULAR BALANCE
As nephrons fail during the course of CKD, each remaining nephron must increase its single nephron excretion of the filtered load of water and solutes. In 1952, Robert Platt1 reasoned that the work of each remaining unit is increased, making an analogy with the remaining workers in a factory putting in overtime when their numbers are reduced by illness. Bricker and colleagues2,3 later noted that the functional capacity of the residual nephrons determines the degree to which homeostasis is preserved, and that maintenance of homeostasis implies preservation (or even enhancement) of normal glomerular and tubular function in the remaining nephrons. Reasoning that globally hypofunctioning nephrons could not maintain homeostasis, these investigators coined the intact nephron hypothesis to explain the preserved ability of the remnant nephrons to excrete the required load of water and solutes.2,3 Each remnant nephron presumably transports water and solutes in proportion to its individual glomerular filtration rate (GFR), whether it is reduced by disease or increased by compensatory hyperfiltration and hypertrophy.
Another essential component of adaptation is maintenance of glomerulotubular balance. For example, in experimental glomerulonephritis, despite a wide range of single nephron (SN)GFR values, proximal fluid reabsorption correlates closely with SNGFR in individual nephrons, so that fractional reabsorption is the same in hypofiltering and hyperfiltering nephrons.4 Presumably, structural changes in the proximal tubule (PT) and the peritubular capillary network act together with alterations in Starling forces to perpetuate glomerulotubular balance.
FUNCTIONAL ADAPTATIONS TO NEPHRON LOSS
Surgical ablation of renal tissue has long been used to mimic the more gradual nephron loss that occurs in CKD. In this experimental model, the reduction of renal mass leads to structural and functional hypertrophy of the remaining nephrons. Remnant nephron hyperfiltration is beneficial in the short term, because it minimizes the decrease in total GFR that would otherwise ensue. However, as will be described, sustained activity of these compensatory forces contributes to a loss of GFR and structural injury in the long term. Most commonly, functional adaptations precede changes in structure, though functional and structural adaptations are closely interrelated, often sharing common pathophysiologic mediators. Adaptive and maladaptive mechanisms will be discussed in parallel, with an emphasis on putative mechanisms or factors underlying transition from adaptation to maladaptation.
FUNCTIONAL ADAPTATIONS
Renal Hemodynamic Adaptations and Maladaptations: Compensatory Hyperfiltration
Remaining nephrons compensate for nephron loss by increased perfusion and filtration rates. Studies in renal transplant donors indicate that within weeks after a nephrectomy,
GFR and the renal plasma flow (RPF) rate in the remaining kidney increase by about 40%, so that GFR is about 70% of the prenephrectomy value.5 In rat models,6,7 vascular resistance is reduced in the afferent and efferent arterioles, allowing an increase in the glomerular capillary plasma flow rate (QA). Because the decrease in afferent arteriolar resistance (RA) is proportionally greater than that in efferent arteriolar resistance (RE), the hydraulic pressure in the glomerular capillary (PGC) increases. Together, glomerular capillary hyperperfusion and hypertension account for the compensatory single nephron hyperfiltration. The magnitude of increase in SNGFR is proportional to the degree of renal mass reduction.8,9 With a uninephrectomy, the increase in SNGFR is largely due to an increase in QA. However, in more extensive renal ablation, substantial increases in PGC occur. The magnitude of the adaptive increase in SNGFR is similar in superficial and juxtamedullary nephrons,10 and the tubuloglomerular feedback mechanism remains intact, with its set point altered in a way that permits hyperfiltration.11
GFR and the renal plasma flow (RPF) rate in the remaining kidney increase by about 40%, so that GFR is about 70% of the prenephrectomy value.5 In rat models,6,7 vascular resistance is reduced in the afferent and efferent arterioles, allowing an increase in the glomerular capillary plasma flow rate (QA). Because the decrease in afferent arteriolar resistance (RA) is proportionally greater than that in efferent arteriolar resistance (RE), the hydraulic pressure in the glomerular capillary (PGC) increases. Together, glomerular capillary hyperperfusion and hypertension account for the compensatory single nephron hyperfiltration. The magnitude of increase in SNGFR is proportional to the degree of renal mass reduction.8,9 With a uninephrectomy, the increase in SNGFR is largely due to an increase in QA. However, in more extensive renal ablation, substantial increases in PGC occur. The magnitude of the adaptive increase in SNGFR is similar in superficial and juxtamedullary nephrons,10 and the tubuloglomerular feedback mechanism remains intact, with its set point altered in a way that permits hyperfiltration.11
Of the glomerular hemodynamic determinants of adaptive hyperfiltration, glomerular capillary hypertension appears to be the crucial cause of eventual structural injury. In a study by Hostetter et al.,8 untreated rats subjected to 85% nephrectomy exhibited elevated SNGFR, due to elevations in PGC and QA. These hemodynamic adaptations were associated with the development of proteinuria and extensive focal and segmental glomerular sclerosis (FSGS).12 When dietary protein restriction was used to blunt the adaptive hyperfiltration, values for SNGFR, QA, and PGC were nearly normalized, and structural injury was limited. Studies with angiotensin converting enzyme inhibitors (ACEIs) helped to clarify the role of specific determinants of hyperfiltration in causing subsequent injury. ACEIs reduce RE and PGC without affecting QA or SNGFR. In rats with renal ablation, selective control of glomerular hypertension is protective, even with persistent hyperfiltration and hyperperfusion. Conversely, antihypertensive therapy that lowers systemic but not intraglomerular pressure may not protect the kidney at risk.9
Loss of renal mass is often accompanied by substantial systemic hypertension8,9 and studies in hypertensive renal disease models have demonstrated the hemodynamic mechanisms of hypertensive injury. Afferent arteriolar vasodilation and an impaired ability to autoregulate in response to changes in perfusion pressure result in enhanced transmission of systemic pressure into the glomerular capillary network and thereby in glomerular capillary hypertension.13
Alterations in Glomerular Permeability
Another index of glomerular function is the ability to restrict passage of macromolecules into the urinary space.14 Permselectivity is characterized by examining the extent to which the glomerular capillary wall discriminates among molecules of different size, charge, and configuration. A uninephrectomy is associated with modest, late development of proteinuria in the rat,15 and in some cases, humans.16 With more extensive renal ablation, proteinuria results from defects in both size selectivity and charge selectivity,12,17 with increased flux through the shunt pathway. As is discussed in the following section, this adaptation is associated with a lesser capability of podocytes to adapt to the loss of nephron mass.
Functional Adaptations: Tubular
Adaptive changes in SNGFR and in proximal tubular reabsorptive capacity are accompanied by mechanisms that enable the excretion of a constant solute load in the face of a dwindling number of functioning nephrons. When this capability is exceeded, the complications of CKD ensue.
As elucidated by Bricker and Fine,3 there are three major patterns of adaptation to advancing CKD (Fig. 75.1), which are reflected by the change in the serum concentration of specific solutes with the fall in GFR. If there is no regulation or adaptation, the plasma concentration increases (curve A of Fig. 75.1). Examples include urea and creatinine, in which the rate of excretion depends on the filtered load, and tubular reabsorption and secretion mechanisms fail to adapt sufficiently to prevent a rise in the serum concentration. Curve B represents regulation with limitation (i.e., maintenance of normal plasma concentration until late stages of CKD); when GFR falls below a critical level, excretion can no longer keep up with intake, and plasma concentration rises. Solutes in this class, such as phosphate and urate, are excreted by filtration and tubular reabsorption, secretion, or both. The third pattern (curve C) is termed complete regulation. Serum concentration is maintained in the normal range until terminal CKD stages; examples include sodium, potassium, and magnesium. For normal serum levels to be maintained, increased single nephron excretion results from altered tubular transport patterns, as discussed in the following. Changes in individual solute handling in CKD have been reviewed extensively elsewhere18 and are briefly summarized here.
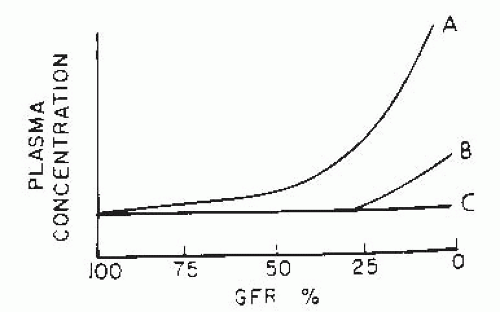 FIGURE 75.1 Patterns of adaptation for different solutes in chronic progressive renal disease. A: No regulation or adaptation. B: Regulation with limitation. C: Complete regulation. See text for discussion. GFR, glomerular filtration rate. (Reprinted from Bricker NS, Fine LG. The renal response to progressive nephron loss. In: Brenner BM, Rector FC Jr, eds. The Kidney. Philadelphia: WB Saunders, 1981:1058, with permission.) |
Sodium Excretion and the Regulation of Extracellular Fluid Volume
Extracellular fluid (ECF) volume is maintained remarkably close to normal until the very late stages of CKD. Although absolute sodium (Na) reabsorption in the PT increases in parallel with the rise in SNGFR, fractional excretion of Na and water increase.19 These changes result in increased Na delivery to the thick ascending limb of Henle and the distal nephron.10
Chronic adaptations enable the maintenance of normal Na balance until the late stages, though the natriuretic response to a large Na challenge is impaired.20 Natriuresis after Na loading is reduced less than is GFR, so acute volume expansion causes a greater increase in fractional Na excretion in uremic animals than in normal controls, a “magnification” phenomenon.20,21 In advanced CKD, the ability to conserve Na is also compromised, such that most patients are unable to lower Na excretion below 20 to 30 mEq per day (a “salt floor”).3,22 With high Na intakes, the smaller number of functioning nephrons may be unable to increase Na excretion enough to maintain Na balance, and thus may be termed as having a low salt ceiling. As CKD progresses, the distance between floor and ceiling increases, and the maintenance of Na balance becomes more difficult,3 though slowly reducing Na intake may lead to more efficient reductions in Na excretion.23
Early investigators postulated that decreased aldosterone formation or effect might be involved in the adaptive changes in Na handling. However, observations that serum aldosterone levels are normal or high, that responsiveness to aldosterone antagonist therapy is present,24 and that exogenous mineralocorticoid therapy does not cause Na retention in CKD patients19,25 have cast doubt on a major role for this mechanism. Later, the discovery of atrial natriuretic peptide (ANP) turned attention toward that hormone as a mediator of Na excretion in CKD. Rats with renal ablation exhibit increased ANP levels, which are related to dietary Na intake and fractional Na excretion.26 Increased ANP levels in CKD patients have been related to increased blood volume and to increased blood pressure.27 Interpretation of these studies is complicated by the presence of heart failure and errors in the plasma ANP assay caused by related peptides that are retained in CKD. However, studies with an ANP antagonist confirm that increased ANP levels contribute to increased fractional Na excretion in experimental CKD28 and giving a monoclonal anti-ANP antibody prevents the postnephrectomy diuresis in rats by blunting both proximal and distal tubular Na reabsorption.29
Hypertension may contribute to increased fractional Na excretion in CKD, as suggested by Guyton’s group.30 According to this view, the maintenance of constant Na intake in the setting of fewer nephrons leads to Na retention and expansion of ECF and blood volumes; the consequent higher blood pressure in turn causes a higher fractional Na excretion. However, blood pressure is higher in CKD patients than in normal subjects whose blood volumes have been increased by salt loading, and reducing dietary salt intake does not consistently prevent hypertension in rats with renal ablation.31 Thus, blood pressure alone is not the sole factor of regulating Na excretion in CKD.
The molecular basis for Na transport after a reduction in nephron number has been explored. Using microdissected tubule segments of 5/6 nephrectomized rats, Terzi et al.32 found increased Na-K-ATPase activity along the nephron when expressed per unit of nephron length. Enzymatic activity correlated with the degree of tubular hypertrophy. However, when expressed per tubule surface unit, no changes were found, suggesting that the density of pumps remained stable during compensatory tubular hypertrophy. Kwon et al.33 found a decrease in total kidney Na/H exchanger, Na-phosphate cotransporter, and basolateral Na-K-ATPase in remnant kidneys as compared to controls. Densities of these transporters did not increase proportionally to the nephron hypertrophy and GFR. These findings reflected mainly changes in the PT and were associated with increased remnant nephron Na excretion. In contrast, expression of bumetanide-sensitive channels in thick ascending limb and thiazide-sensitive channels in distal tubules was increased, and Na-K-ATPase expression was maintained in these segments. These changes indicate compensatory increases in distal segments in CKD, partly because of elevated vasopressin and aldosterone levels. In contrast to rats with extensive renal ablation, studies in uninephrectomized rats have shown upregulation and activation of NHE-3 (Na/H exchanger). Moreover, the inhibition of NHE-3 was associated with the inhibition of compensatory growth in the remnant kidney.34
Potassium Homeostasis
The kidney retains the ability to maintain potassium (K) homeostasis and normal serum K levels until very late stages of CKD. K secretion per nephron increases, and experimentally, the fractional excretion of K may exceed 100% of the filtered load. Similar to Na handling, there is a significant inverse correlation between the fractional excretion of K and GFR. The major factors responsible for increased K excretion per nephron appear to be the elevation of plasma and intracellular K concentrations following K ingestion, particularly early in the course; later, an adaptive tubule process also augments K secretion.35 In uninephrectomized rats, K secretion occurs within hours after a nephrectomy and is mediated by amiloride-sensitive channels.36 In addition, reduced K reabsorption by the loop of Henle may facilitate the excretion of acute potassium loads in CKD.37
Following the ingestion of a K load, serum K increases by about the same increment in CKD patients and in normal subjects, inducing an increase in distal K secretion. When factored for GFR, the kaliuresis in patients with moderate CKD is the same as in normal subjects.38 However, because the CKD patient excretes K more slowly than in the normal subject, there is prolonged elevation of serum K following an oral load.
Later in the course, distal tubular adaptations— specifically, increased activity of Na-K-ATPase and basolateral surface area in principal cells of the cortical collecting duct—promote K excretion.39 In addition, as CKD progresses, intestinal K excretion also increases in concert with increased colonic Na-K-ATPase activity40,41 and potassium permeability.42 The administration of spironolactone to patients with CKD may result in dangerous hyperkalemia. Thus, it appears that adequate aldosterone levels are required to facilitate increased K secretion per nephron, and that hyperkalemia may occur earlier in CKD patients with low plasma aldosterone levels.43
Water Homeostasis
The capacity to generate solute-free water is remarkably well maintained in moderate CKD.3,44,45 However, water excretory capacity is impaired, as reflected in a reduction in the minimum attainable urine osmolality. In contrast to the somewhat preserved diluting mechanisms, concentrating ability starts to fail relatively early in CKD, resulting in decreased fractional reabsorption of solute-free water. In CKD, severe dehydration is usually avoided because intact thirst mechanisms allow the patient to compensate for the urinary water losses. Accordingly, nocturia is the predominant symptom.
Urinary concentration requires the maintenance of hypertonicity of the medullary interstitium and normal water transport across distal nephron segments in response to antidiuretic hormone (ADH). The maintenance of medullary interstitial hypertonicity in turn requires structural preservation of the countercurrent system. Part of the urinary concentrating defect in CKD may be attributed to the high solute load imposed on each nephron. However, disruption of medullary architecture in tubulointerstitial diseases may cause disproportionate impairment of concentrating ability.46 The presence of a concentration defect despite elevated plasma ADH levels in CKD suggests a distal tubular defect. Limited ADH responsiveness may be due to two factors. First, ADHstimulated adenylate cyclase activity and water permeability in the distal nephron may be impaired.47,48 Also, increased tubular flow rates may limit the fraction of water that can be reabsorbed by the distal nephron in response to ADH. Teitelbaum and McGuinness49 described the absence of ADH V2 receptor mRNA in the inner medulla of rats with CKD. Kwon et al.50 studied protein expression of aquaporins (AQP) in rat remnant kidneys. AQP1 is found mainly in the proximal tubule and in the descending limb of the loop of Henle, whereas AQP2 and 3 localize in the apical and basolateral membranes of the collecting ducts, respectively. All three channels were markedly reduced in remnant kidneys. Furthermore, decreased AQP expression was resistant to treatment with ADH.50 Downregulation of the Na cotransporter rat bumetanide-sensitive cotransporter (rBSCl) has also been reported.51 Several molecular mechanisms involving AQP in CKD have been suggested (reviewed in Holmes52).
Acid-Base Homeostasis
Metabolic acidosis is characteristic of CKD, due to reduced renal ability to excrete acid.53,54 Early in the course, hydrogen balance is maintained by increased ammonium excretion per functioning nephron.53,54,55 Later, this adaptation proves insufficient and acidosis is maintained due to reduced ammonia synthetic capacity. Ammonium excretion per total GFR rises to three to four times normal,56 but the increase is insufficient to counteract the reduced nephron number. Impaired ammonia excretion was originally attributed to the impairment of countercurrent mechanisms, which were thought to increase ammonium concentration in the medulla and facilitate “trapping” of ammonia by acidified luminal fluid in the collecting duct. However, it is now recognized that ammonia enters tubule fluid by active secretion as well as by trapping.
Renal acid excretion also requires the reabsorption of filtered bicarbonate and the generation of a large hydrogen ion gradient in the distal nephron. Following a uninephrectomy, the stimulation of PT bicarbonate reabsorption occurs, as a result of a doubled transport rate. An increase in Na/H exchange contributes to this phenomenon.57 There may be slight increases in the fractional reabsorption of bicarbonate58 and studies of proximal tubule brush-border vesicles from rats with renal ablation have shown increases in Vmax of the Na-H antiporter.59 However, clinically, a decrease in bicarbonate reabsorptive ability develops,60 corresponding to findings of reduced NHE-3 in the proximal tubules of rat remnant kidneys.33 In severe CKD, the threshold for bicarbonate reabsorption may also be reduced.61
Distal acidification is generally better maintained than proximal bicarbonate reabsorption in CKD, except in patients with distal renal tubular acidosis. However, CKD patients are unable to lower urinary pH as well as normal subjects with experimental acidemia,53 and so a relative decrease in distal hydrogen ion pump capacity may also contribute to acidosis. Failure to attain normal minimal urinary pH prevents optimal titration of nonammonia buffers and thus reduces the excretion of the titratable acid. Dietary phosphate restriction may further contribute to the reduced excretion of titratable acid in CKD.
Phosphate Homeostasis
Abnormalities of phosphate and calcium metabolism and their contributions to metabolic bone disease are discussed elsewhere in this book. Intrarenal adaptations that contribute to calcium and phosphate homeostasis in CKD are discussed here. A progressive increase in fractional phosphate excretion maintains phosphate balance in early CKD.62 Later in the course, phosphate excretion is maintained by a further increase in the fractional excretion, along with increased serum phosphate levels. Slatopolsky et al.62,63,64 suggested that increased parathyroid hormone (PTH) caused the increase in fractional phosphate excretion. In dogs with renal ablation, the fractional excretion of phosphorus increased and the magnitude of the increased fractional excretion
correlated with the magnitude of the increase in circulating PTH levels. Restricting phosphate intake prevented increases in both PTH levels and fractional phosphate excretion. These observations formed the basis for Bricker’s “trade-off hypothesis,”65 postulating that some of the major stigmata of uremia may occur as indirect consequences of the adaptations in the nephron function.3 According to this hypothesis, hyperparathyroidism is the biologic price paid to maintain the excretion of a constant dietary phosphate load when the nephron number is reduced. With each decrement in GFR, a transient period of phosphate retention and decreased plasma calcium would stimulate PTH synthesis and secretion, and the increased PTH activity would act to partially restore phosphate balance by augmented phosphate excretion.66 This adverse trade-off could be avoided by reducing phosphate intake as renal function declined.
correlated with the magnitude of the increase in circulating PTH levels. Restricting phosphate intake prevented increases in both PTH levels and fractional phosphate excretion. These observations formed the basis for Bricker’s “trade-off hypothesis,”65 postulating that some of the major stigmata of uremia may occur as indirect consequences of the adaptations in the nephron function.3 According to this hypothesis, hyperparathyroidism is the biologic price paid to maintain the excretion of a constant dietary phosphate load when the nephron number is reduced. With each decrement in GFR, a transient period of phosphate retention and decreased plasma calcium would stimulate PTH synthesis and secretion, and the increased PTH activity would act to partially restore phosphate balance by augmented phosphate excretion.66 This adverse trade-off could be avoided by reducing phosphate intake as renal function declined.
However, some studies indicate that the increased fractional phosphate excretion does not depend on an increase in PTH levels or tubule responsiveness to PTH.67 These data suggest that the increased fractional excretion is achieved via decreased phosphate reabsorption in the proximal nephron, as occurs in intact animals fed a high phosphate diet.68 Phosphate uptake per unit tubule mass is reduced in the proximal nephron segments isolated from uremic rabbits, and sodiumphosphate cotransport activity is reduced in brush-border membrane vesicles from uremic dogs69 and rats33, but these reductions do not fully account for the reduction in proximal phosphate reabsorption observed in vivo. Although reduced proximal reabsorption accounts for most of the increased fractional phosphate excretion, there is also evidence of altered distal phosphate transport (or reabsorption).68 Nevertheless, the principal tenets of the intact nephron hypothesis continue to inform studies of adaptation in CKD.70
Calcium Homeostasis
As CKD advances, the production of active vitamin D [1,25(OH2)D3] is impaired, leading to reduced intestinal calcium reabsorption and renal calcium excretion; the fractional calcium excretion then increases.71 The mechanism responsible for the increased fractional calcium excretion in advanced CKD is unclear. Possible factors include acidosis, the suppression of vitamin D production, increased distal nephron flow rates, and ECF volume expansion. Animal studies suggest that the increase in remnant nephron calcium excretion associated with ECF expansion may be mediated by ANP.72
STRUCTURAL ADAPTATIONS
Renal hypertrophy is an early development in the adaptive response to loss of renal mass.73,74,75 Enlargement is due primarily to growth of the proximal convoluted tubules,76 resulting in the disproportionate enlargement of the cortex in comparison to the medulla. In 1917, Addis77 reasoned that the excretion of urea required energy, and that renal enlargement was a reflection of the need for added renal “work.” Although he subsequently disproved his own hypothesis with regard to urea, the notion that the kidney grows in response to the added workload remains. Despite ingenious experimentation over the years, the question of whether hyperfiltration drives hypertrophy, or vice versa, remains incompletely answered. However, new understandings of the mechanisms and consequences of mechanotransduction are helping to elucidate this question.
Compensatory Renal Hypertrophy
Structural Changes in Individual Renal Compartments
Glomerular Hypertrophy Glomeruli also undergo progressive enlargement. A uninephrectomy in the rat increases glomerular tuft volume (VG) by up to 75%.78 In more exuberant hypertrophic states, as occur with extensive ablation or mineralocorticoid-salt hypertension, the degree of VG enlargement is even greater.78,79,80 Increased VG does not necessarily parallel increased whole kidney size; VG may continue to increase after renal growth slows. Serial structure-function studies in the rat have shown that VG and SNGFR increase in parallel after uninephrectomy.79,80 Morphometric studies demonstrate an increase in the total volume occupied by cellular constituents in the remnant glomeruli of nephrectomized rats.79,80,81,82,83,84,85,86 Overall, the glomerular fractions occupied by different structural components remain constant as VG enlarges, at least in the early phases of adaptation. However, long-term adaptation may follow a different pattern, with the mesangial volume fraction increasing only later in the course.81
An early study found a prominent increase in the number of glomerular endothelial, mesangial, and epithelial cells following a uninephrectomy in very young rats.84 Later studies confirmed increased mesangial cellularity.86 A proliferative response involving glomerular endothelial cells is apparent,87,88,89 as is an increase in endothelial cell volume.87
Increases in glomerular capillary length and radius occur, such that capillary surface area increases.81,86 Afferent and efferent arteriolar diameters also increase.86 Morphometric measurements have been used to estimate the filtering capacity of the enlarged remnant glomeruli. The glomerular capillary ultrafiltration coefficient (Kf) is the product of the surface area available for filtration (S) and the hydraulic permeability of the glomerular capillary wall (k). It is not clear which anatomic boundary constitutes the surface corresponding to S. As estimated by measuring the glomerular capillary area in direct apposition to epithelial foot processes, S increases following a nephrectomy, albeit to a slightly lesser degree than total glomerular volume.79 However, most functional studies have not found an increase in Kf of remnant glomeruli after extensive renal ablation.8,9
It is conceivable that k is reduced, because a decrease in S cannot be involved to explain the fall in Kf. In theory, an increase in average foot-process width would cause a decrease
in the length of filtration slit overlying each unit area of peripheral capillary surface and thereby could decrease k in remnant glomeruli. In fact, morphometric studies have revealed an increase in the average width of epithelial cell foot processes in rats subjected to extensive renal ablation.80 Alternatively, it may be that the filtering surface estimated by morphologic techniques in remnant glomeruli does not represent effective area available for filtration in vivo. Theoretical studies suggest that much of the glomerular capillary network is relatively underperfused in rats with extensive renal ablation.90 No increase in S was found following a uninephrectomy in rats when the infusion of glomerular basement membrane antibody was used to estimate capillary surface area in vivo.91 Alternatively, it may be that the decrease in Kf in remnant glomeruli is more functional than structural, at least early on, because ACEI therapy raises Kf to supranormal levels.9
in the length of filtration slit overlying each unit area of peripheral capillary surface and thereby could decrease k in remnant glomeruli. In fact, morphometric studies have revealed an increase in the average width of epithelial cell foot processes in rats subjected to extensive renal ablation.80 Alternatively, it may be that the filtering surface estimated by morphologic techniques in remnant glomeruli does not represent effective area available for filtration in vivo. Theoretical studies suggest that much of the glomerular capillary network is relatively underperfused in rats with extensive renal ablation.90 No increase in S was found following a uninephrectomy in rats when the infusion of glomerular basement membrane antibody was used to estimate capillary surface area in vivo.91 Alternatively, it may be that the decrease in Kf in remnant glomeruli is more functional than structural, at least early on, because ACEI therapy raises Kf to supranormal levels.9
Compensatory glomerular hypertrophy has been proposed as a trigger of FSGS as well. Many models show a strong association between increased VG and the development of proteinuria and FSGS.31,78,79,92 Conversely, there may be a protective effect of low VG.93 Miller et al.94 compared the rate of development of proteinuria and FSGS in normal and uninephrectomized rats treated with a pressor dose of angiotensin (Ang) II, which increases PGC. Despite the fact that the Ang II dose was halved in uninephrectomized rats, these rats demonstrated a markedly faster development of renal injury. There were no differences in PGC between those groups; however, VG was increased in the uninephrectomized animals.
The combination of increases in both glomerular capillary intraluminal pressure and capillary radius exerts increased tension on the glomerular capillary wall (following Laplace law), thus contributing to disruption of capillary wall integrity, the induction of mechanical stress-induced signaling, the activation of local humoral systems, and the podocyte changes discussed in the following. Normalizing PGC may limit, but does not prevent, glomerular hypertrophy following renal ablation. Studies comparing two models of renal ablation showed differences in systemic and glomerular blood pressure, the expression of prosclerotic factors, and the rate of development of renal injury, but similar degrees of glomerular hypertrophy.95 These studies indicate that the protective effect of reducing PGC cannot be solely attributed to reduction in VG.
Podocyte Biology after Nephron Loss. The potential additive deleterious effects of glomerular capillary hypertension and glomerular enlargement also suggest that injury may be mediated by effects on the glomerular visceral epithelial cells79,92 and their lower ability to adapt. Adult podocytes possess diminished ability to divide in response to stressful stimuli.96,97 Podocytes undergo exaggerated stress as glomeruli enlarge, resulting in their dysfunction and possibly destruction, in the pathogenesis of FSGS.79,82
Though unable to replicate, podocytes are able to undergo hypertrophy in response to a loss of renal mass and glomerular enlargement.82,83 As reviewed by Kriz and LeHir,97 glomerular enlargement without podocyte replication leads to structural changes including foot-process effacement, cell-body attenuation, pseudocyst formation, accumulation of absorption droplets, and finally, detachment. The loss of podocytes then triggers the onset of FSGS, as is further discussed.
Mechanisms of Renal Hypertrophy
General Principles: Hypertrophy Versus Hyperplasia
The term compensatory hypertrophy has been used to describe the aggregate changes in nephron structure, including both cellular hypertrophy and hyperplasia, which follow a loss of renal mass (reviewed in Fine and Norman,98 Wolf,99 Terzi et al.100). Renal cells react to physical, biochemical, and humoral stimuli imposed by reduced nephron number with coordinate activation of a variety of signaling pathways and changes in expression of their molecular components, with a resultant induction of genes encoding proteins involved in structural adaptive and maladaptive responses.
Hyperplasia is a result of an increase in cell number associated with DNA replication and cell division, whereas hypertrophy is defined as cell enlargement due to increases in protein and RNA content without DNA replication.75 Both processes are involved in renal compensatory growth. During hyperplasia, cells progress through the whole cell cycle; hypertrophy occurs when the cells are engaged in the cycle but cannot progress to later stages. An interaction of the cell with growth factors that modulate the activity of cell cycle regulators then determines whether the cell will engage in hyperplasia or hypertrophy.
The Role of Physical Forces
Cortes and coworkers101,102 showed that glomeruli can enlarge as perfusion pressure rises. In the remnant kidney, the transmission of pressure fluctuations into the glomerular capillary tuft is not prevented by myogenic autoregulatory control. This results in glomerular distension and increased VG. Glomerular compliance is determined by capillary wall tension, size of the glomerulus, and glomerular stiffness, and is increased in remnant kidneys. This process is independent of humoral or biochemical factors, at least initially. Indeed, an increase in glomerular capillary radius is the earliest morphologic finding after a uninephrectomy.82,83
Increased glomerular capillary pressures and/or plasma flow rates alter the growth and activity of glomerular component cells, inducing the expression of cytokines and other mediators, which then stimulate mesangial matrix production and promote structural injury. Hemodynamic physical forces, such as shear stress or changes in blood flow, are well recognized to influence activity of endothelial and vascular smooth muscle cells (VSMC). Mechanical stress imposed on renal cells by enhanced capillary and tubular flows and pressures triggers a variety of physiologic
and pathophysiologic responses. A key component of these responses, representing the actual link between mechanical stresses and growth in glomeruli and tubules, is stress-induced signaling or mechanotransduction. Intrarenal mechanotransduction has been demonstrated using in vitro systems capable of simulating stretch or pulsatile stresses imposed upon endothelial, mesangial, and tubular cells, or podocytes.
and pathophysiologic responses. A key component of these responses, representing the actual link between mechanical stresses and growth in glomeruli and tubules, is stress-induced signaling or mechanotransduction. Intrarenal mechanotransduction has been demonstrated using in vitro systems capable of simulating stretch or pulsatile stresses imposed upon endothelial, mesangial, and tubular cells, or podocytes.
Principles of Mechanotransduction
Physical stimuli are sensed by cells and transmitted through intracellular signal transduction pathways to cytosolic effectors or to the nucleus, resulting in the induction of inappropriate genes.103,104,105 The responses to mechanical stress lead to both adaptive and maladaptive processes following the nephron loss.
A spectrum of molecules has been implicated in mechanosensing; mechanical stress may directly perturb the cell surface and alter conformation of receptors and their immediate downstream effectors such as G proteins, guanine nucleotide exchange factors, and small GTPases of the Rho family, thereby initiating signaling pathways. The role of integrins, cadherin junctional complexes, and the cytoskeleton in mechanosensing has also been suggested by multiple lines of evidence.103,104,105,106,107 The extracellular matrix (ECM) protein organization is sensed by integrins, transmembrane receptors that mediate cell attachment to ECM proteins. Integrins act as mediators of cell adhesion, but can also transmit extracellular stimuli into intracellular signaling events. In the vascular system, the transient receptor potential (TRP) ion channel superfamily has also been implicated in mechanosensing, as well as in stress-induced Ca2+ influx and vascular functional alterations,108 but their role in renal responses to nephron loss remains unknown. Also, new caveolae are formed in endothelial cells in response to shear stress, and act as mechanosensors resulting in ras-raf-extracellular signal-related kinase (ERK) and Akt kinase activation.109
Mechanosensing molecules transduce signals to downstream kinases and other signaling mediators. In vascular and renal cells, mechanotransduction involves the activation of the mitogen-activated protein kinase (MAPK) family.110 These serine/threonine kinases transduce signals in response to multiple agonists acting through GFRs with intrinsic tyrosine kinases, G protein-coupled receptors, via nonreceptor tyrosine kinases, and cellular stresses. ERKs have been implicated in mitogenic cellular responses, whereas c-Jun Nterminal kinase (JNK) and p38 activation is associated with inflammatory cytokine action, oxidative stress, prosclerotic actions, production of ECM, and apoptosis.111,112 Activated MAPKs can translocate to the nucleus and can lead to phosphorylation and activation of transcriptional factors (which follow) or phosphorylate their cytosolic substrates.113
Protein kinase C (PKC) is another major family of serine/threonine kinases that are active in mechanotransduction. PKC are typically activated by lipid-derived second messengers, such as diacylglycerol or phospholipids. PKC has a number of downstream targets involved in hypertrophic signaling and the production of vasoactive factors.114 PKC has also been shown to contribute to shear stress-induced MAPK activation.
Phosphatidylinositol-3 kinase (PI-3K), another multifactional kinase activated by upstream G proteins, is stimulated by shear stress111 and contributes to shear stress-induced JNK activation in endothelial cells. PI3K is also involved in endothelial mechanotransduction sensed by integrins.112 Akt kinase (protein kinase B) is a major downstream target of PI-3K. Akt is a potent survival kinase, transducing signals leading to the inhibition of apoptosis promoting cell proliferation and cell cycle regulation. Akt kinase may also transduce potentially protective signals, such as shear stress-induced generation of nitric oxide.115
Rho associated kinases, the effectors of the Rho family of small G proteins that act as possible mechanosensors, have been implicated in mediating cell contraction, adhesion, migration, cytoskeleton organization, and proliferation, and have been identified as major players in the pathophysiology of CKD.116,117
Activation of growth promoting systems leads to the upregulation of transcription factors such as activator-protein 1 (AP-1), a transcription factor formed by the heterodimerization of fos and jun proteins, or nuclear factor-κB (NF-κB), and early response genes or proto-oncogenes, the protein products of which regulate the transcriptional control of large numbers of other genes, which are early steps in cell proliferation and differentiation evoked by stress signaling, mitogens, and growth factors.
The Impact of Mechanical Stress
Expansion of the glomerular capillaries and stretching of the mesangium in response to hypertension might be a force that translates high PGC into increased mesangial matrix formation. In microperfused rat glomeruli, increased PGC is associated with increased VG; in cultured mesangial cells, cyclic stretching results in the enhanced synthesis of protein, total collagen, collagen IV, collagen I, laminin, fibronectin, and transforming growth factor (TGF)-β.101 Additionally, growing mesangial cells under pulsatile conditions stimulates the Ang II receptor; angiotensinogen mRNA levels118 and protein kinase C, calcium influx, and proto-oncogene expression119; as well as altered extracellular matrix protein processing enzymes.120,121 In vivo, Shankland et al.122 observed that the increased PGC induced by uninephrectomy in spontaneously hypertensive rats (SHR) was associated with the glomerular expression of TGF-β and platelet-derived growth factor (PDGF). Normalization of PGC with an ACEI decreased glomerular TGF-β and PDGF. Similarly, Griffin et al.95 compared PGC in relation to glomerular TGF-β and PDGF expression between the excision and infarction models of renal ablation. The rats subjected to renal infarction demonstrated higher systemic and glomerular pressures, and a markedly higher expression of TGF-β and PDGF mRNA in glomeruli.
Ingram and coworkers110,114 found stretch-induced activation of p42/44, JNK, and p38 in mesangial cells. These changes resulted in the activation of the AP-1 family, and were in part Ca2+ and PKC dependent. Stretch-induced activation of p38 MAPK in mesangial cells leads to increased production of TGF-β.111 Of note, Ohashi et al.112 found that the inhibition of enhanced renal p38 MAPK activity in rats with renal ablation leads to the acceleration of renal injury, with more proteinuria, FSGS, and interstitial fibrosis, but less tubular cell apoptosis. p38 Inhibition was associated with ERK 1/2 activation, a possible factor explaining the worsening FSGS. Thus, activation of p38 in remnant kidneys might play a protective role associated with inhibitory actions on ERKs signaling.
AP-1 and NF-κB are increased in remnant kidneys and downregulated by nephroprotective treatment.123,124 Mirza et al.125 reported an important function of NF-κB in regulating of the enzyme transglutaminase, which is an activator of latent TGF-β. Transglutaminase also cross-links matrix proteins, possibly contributing to interstitial fibrosis in the remnant kidney model.126 The activation of NF-κB may have an important role in mediating cortical interstitial monocyte infiltration and tubular injury in proteinuric tubulointerstitial inflammation.127,128 Studies of expression of early response genes in the remnant kidney following a uninephrectomy have been inconsistent. For example, the renal activity of c-fos, c-myc, c-egr1, c-jun, and c-H-ras following a uninephrectomy has been found to increase in some studies, but not in others.129,130,131,132 Importantly, however, these studies suggest that proto-oncogene activation after uninephrectomy is modest or absent, consistent with the low risk of deterioration of GFR in this modest degree of nephron loss.
Cell Cycle Regulation as an Important Determinant of Structural Response to Nephron Loss
Genes induced by nephron loss include those coding for cell cycle regulatory proteins.133 Positive regulation (the stimulation of transition from quiescent cell phase to ultimate cell division by mitosis) is carried out by cyclins and their partner molecules, cyclin-dependent kinases (CDK). Although CDK are constitutively expressed, cyclins are transcriptionally regulated and their levels are increased by specific mitogens such as growth factors. Negative regulation of the cell cycle is accomplished by CDK inhibitors, which bind to cyclin-CDK complexes and inhibit their activity. In addition to the preceding processes, the cells also engage in apoptosis. The total number of cells in a particular organ reflects the balance between proliferation and apoptosis. Apoptotic cells exit from the cell cycle. The initiation of apoptosis also is regulated by cyclin-CDK complexes, and the progression of the cell into mitosis or apoptosis seems to be determined by the level of CDK inhibitor p27.133
Liu and Preisig134 studied renal tubular cell cycle regulation after a uninephrectomy and the resulting compensatory hypertrophy. In both rats and mice, compensatory PT growth after the uninephrectomy was hypertrophic and was associated with a cell cycle-dependent mechanism. The development of hypertrophy required that the cells enter the G1 phase and initiate the events of this phase, such as increased protein synthesis, increased cdk4/cyclin D kinase activity, and maintaining retinoblastoma protein in the hypophosphorylated state. However, the cells did not progress to the S phase, where DNA synthesis occurs. Unlike the cdk4/cyclin D activation, there was insufficient or absent activation of cdk2/cyclin E, preventing the progression of cell cycle into the S phase, resulting in hypertrophy instead of hyperplasia. This pattern of differential activation of cdk4/cyclin D and cdk2/cyclin E may reflect the previously mentioned differences in the expression and activities of p21 and p27. Compensatory hypertrophy after renal ablation is associated with cyclin E and CDK2 expression coinciding with the early proliferative response.135
CDK inhibitors also play a key role. These molecules are regulated by factors that have been implicated in ablation nephropathy, including Ang II and TGF-β.136,137 p21 knockout mice subjected to renal ablation are resistant to the development of FSGS, as compared to wild-type animals.138 In the absence of the p21 gene, the growth response in the remnant kidney is relatively more hyperplastic than hypertrophic. The authors alluded to a proposal, made by Goss139 45 years ago, suggesting that when an organ accommodates increased work by hypertrophy rather than hyperplasia, it is at a serious physiologic disadvantage and is more likely to undergo regression of structure and function.
Mechanotransduction in Podocytes and Its Consequences for Glomerular Injury. The additive deleterious effects of glomerular capillary hypertension and glomerular enlargement have also prompted speculation that injury may be mediated by detrimental effects on the glomerular visceral epithelial cells.79,83,92 Podocytes undergo exaggerated stress as glomeruli enlarge, resulting in their dysfunction and possibly destruction.79,82 Adult podocytes possess a diminished ability to divide in response to stressful stimuli96,97; though unable to replicate, they are able to undergo hypertrophy. As reviewed by Kriz and LeHir,97 glomerular enlargement without podocyte replication leads to structural changes including foot-process effacement, cell-body attenuation, pseudocyst formation, accumulation of absorption droplets, and finally, detachment. The loss of podocytes then triggers the onset of FSGS, as is discussed below.
Mechanical stress induces a unique reorganization of the actin cytoskeleton in podocytes.115 The F-actin reorganization in response to mechanical stress depends on Ca2+ influx and Rho kinase. A dynamic cytoskeleton allows podocytes to withstand significant mechanical stress with elevation of PGC. Podocytes respond to fluid shear stress with a changed expression of focal contact markers and downregulation of ZO-1 followed by a loss of cell-cell contacts.140 That process leads to an intermediate adhesive state, which
may be a promoter of podocyte detachment. That study also demonstrated that the activation of specific tyrosine kinases is required for the podocytes to withstand increased fluid shear stress.140
may be a promoter of podocyte detachment. That study also demonstrated that the activation of specific tyrosine kinases is required for the podocytes to withstand increased fluid shear stress.140
Stretch-induced p38 in podocytes is associated with an enhanced prostanoid production via cyclooxygenase-2 (COX-2), an increased expression of the EP4 prostanoid receptor, and consequent alterations in podocyte cytoskeletal dynamics that could compromise filtration barrier function under conditions of increased PGC.141 In CKD, inhibitors have been implicated in mechanical stretch-induced podocyte hypertrophy.142 Stretch reduces cell-cycle progression in wild-type and p27 knockout mice and induces hypertrophy, whereas hypertrophy is not induced in single p21 and double p21/p27 knockout podocytes. Stretch-induced hypertrophy is inhibited by blocking ERK 1/2 or Akt, but not p38.
Integrins are essential for podocyte adhesion to the glomerular basement membrane and therefore to the integrity of glomerular filter. Every integrin binds to a restricted number of ECM ligands. The laminin binding integrin α3β is the most highly expressed integrin on podocytes, and the key integrin mediating podocyte adhesion to the GBM.143 Podocyte adhesion to the GBM via integrin α3β1 is enhanced by its interaction with the tetraspanin protein CD151,144,145 which regulates the tightness of integrin-dependent adhesion, cell morphology, and cell migration. The role of mechanical stress in podocyte injury and the consequent development of FSGS is further supported by evidence in CD151 knockout mice.144 These observations have suggested that podocyte adhesion to the GBM promoted by CD151 is required to prevent the development of FSGS under conditions of high PGC, and that ACEIs induce their therapeutic effects, at least in part by limiting podocyte detachment from the GBM.
Tubular Structural Adaptations
Quantitatively, the proximal convoluted tubule enlarges by approximately 15% in luminal and outside diameter and by 35% in length after the uninephrectomy in the rat,76 and PT enlargement is proportional to the extent of the nephron number reduction.146 The increased tubule size follows the increase in fluid reabsorption by segments of the isolated proximal straight tubule.147 Later in the course, the increased reabsorptive rate approximates the increase in PT size and protein content.148 The distal convoluted tubule enlarges by approximately 10% in the luminal and outside diameter by 17% in length in the rat.76 In the distal convoluted tubules and collecting ducts, the cross-sectional area of both lumen and epithelium is increased, but to a lesser degree.76 Tubular cells do not enlarge symmetrically; enlargement of basolateral portions of cells is more prominent as compared to the luminal surface.149
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


