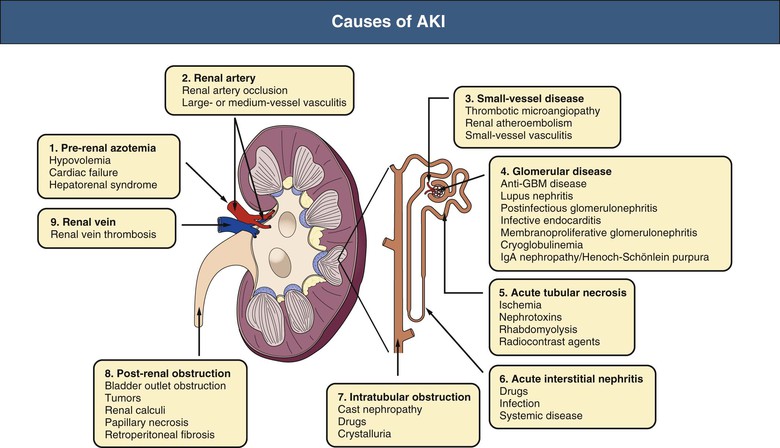
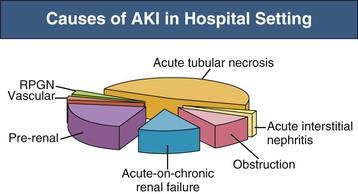
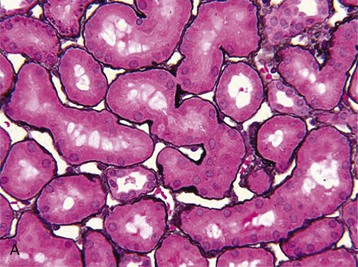
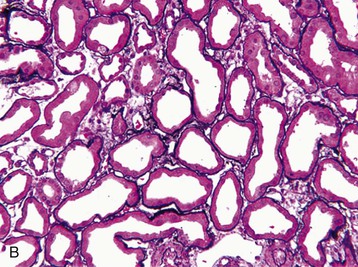
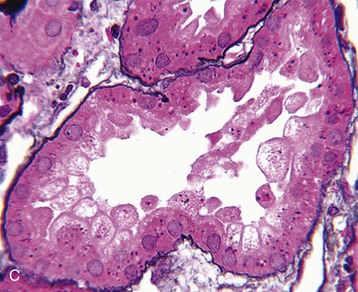
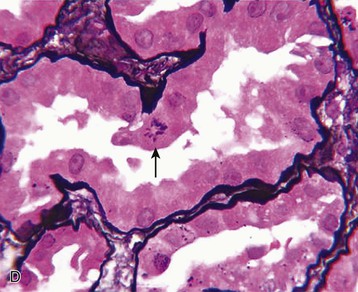
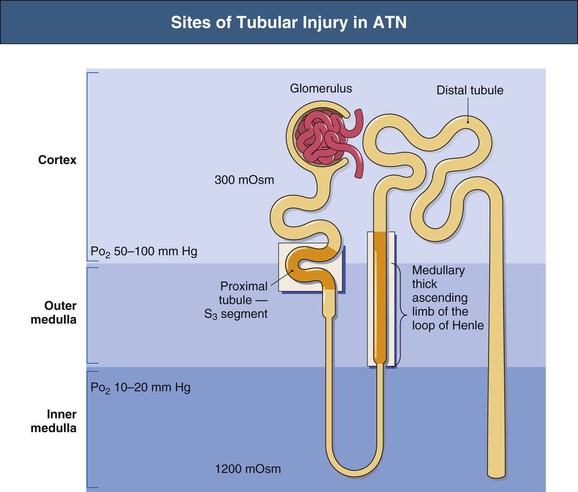
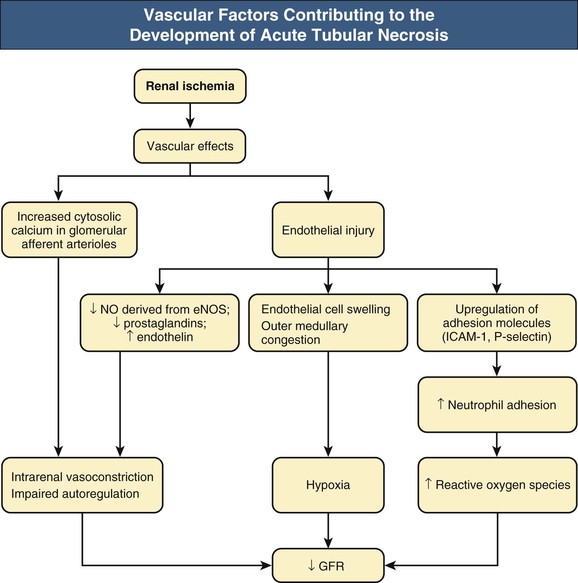
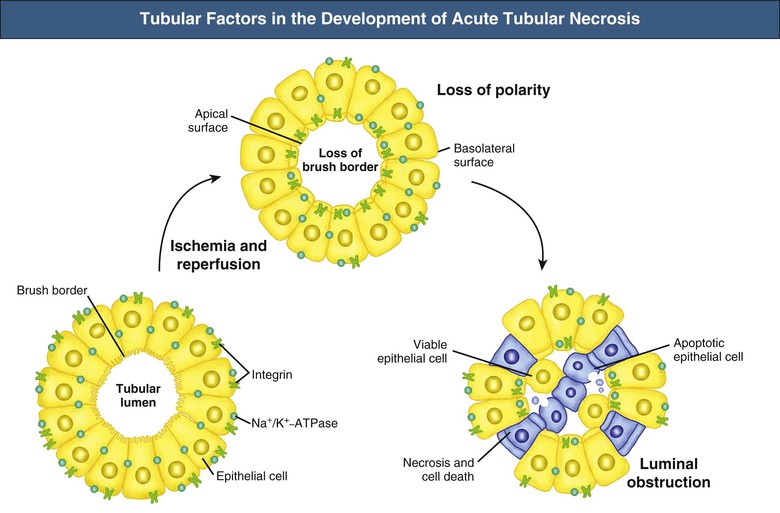
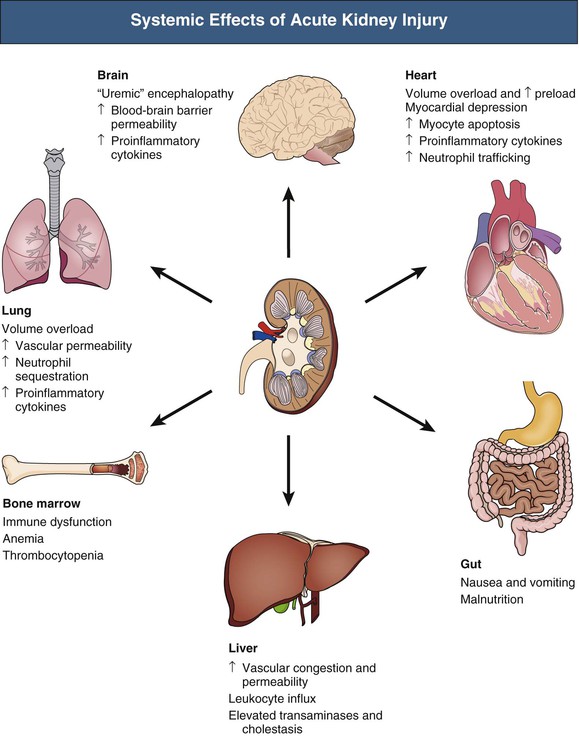
J. Ashley Jefferson, Joshua M. Thurman, Robert W. Schrier Acute kidney injury (AKI) is a clinical syndrome denoted by an abrupt decline in glomerular filtration rate (GFR) sufficient to decrease the elimination of nitrogenous waste products (urea and creatinine) and other uremic toxins. This has traditionally been referred to as acute renal failure (ARF), but in recent years an effort has been made to implement the term acute kidney injury instead, and to develop a standardized definition of AKI. One proposed definition of AKI, for example, is a decline in kidney function over 48 hours as demonstrated by an increase in serum creatinine of greater than 0.3 mg/dl, an increase in serum creatinine of more than 50%, or the development of oliguria.1 Staging criteria have also been developed based on the magnitude of the rise in serum creatinine and changes in the volume of urine output over 1 week,1 and studies have validated that these staging criteria are of prognostic value (see Table 71-1). Although AKI is defined by a reduced GFR, the underlying cause of the renal impairment is most frequently a result of tubular and vascular factors. AKI can have a broad range of causes, and the differential diagnosis must be considered in a systematic fashion to avoid missing multiple factors that may be contributing to the condition. The traditional paradigm divides AKI into pre-renal, renal, and post-renal causes. Pre-renal uremia may be caused by hypovolemia or a decreased effective arterial volume. Post-renal obstructive renal failure is usually diagnosed by urinary tract dilation on renal ultrasound. Intrinsic renal causes of AKI should be considered under the different anatomic components of the kidney (vascular supply; glomerular, tubular, and interstitial disease; Fig. 69-1). Major extra-renal artery or venous occlusion must also be considered in the differential diagnosis (see Chapter 66). Similarly, disorders of the small intrarenal vasculature can result in AKI (e.g., vasculitis, thrombotic microangiopathy [TMA], malignant hypertension, eclampsia, postpartum states, disseminated intravascular coagulation [DIC], scleroderma; see Chapters 25, 29, 37, 44, and 64). All forms of acute glomerulonephritis (GN) can present as AKI, as can acute inflammation and space-occupying processes of the renal interstitium (e.g., drug-induced, infectious, and autoimmune disorders, leukemia, lymphoma, sarcoidosis). In the hospital setting, pre-renal uremia and acute tubular necrosis (ATN) account for the majority of AKI cases,2,3 often in the setting of AKI superimposed on chronic kidney disease (CKD), so-called “acute-on-chronic renal failure” (Fig. 69-2). The term tubular necrosis is a misnomer because the alterations are not limited to the tubular structures and true cellular necrosis in human ATN is often minimal. However, the term acute tubular necrosis is commonly used in the clinical setting. To make things even more confusing, the terms acute tubular necrosis, acute renal failure, and acute kidney injury are frequently used interchangeably in the literature. The term acute tubular necrosis should be reserved for cases of AKI in which a renal biopsy (if performed) shows the characteristic changes of tubular cell injury, and for patients with findings of tubular injury (such as renal tubular epithelial cells in the urine sediment) in an appropriate clinical setting (Fig. 69-3). There are also significant geographic differences in the causes of AKI; the spectrum of causes in tropical countries is described in Chapter 70. Impaired renal perfusion with a resultant fall in glomerular capillary filtration pressure is a common cause of AKI. In this setting, tubular function is typically normal, renal reabsorption of sodium and water is increased, and consequently urine chemistries reveal a low urine sodium (<20 mmol/l) and a concentrated urine (urine osmolality >500 mOsm/kg) as long as no loop diuretic has been administered. A marked reduction in renal perfusion may overwhelm autoregulation and precipitate an acute fall in GFR. With lesser degrees of renal hypoperfusion, glomerular filtration pressures and GFR are maintained by afferent arteriolar vasodilation (mediated by vasodilatory eicosanoids) and efferent arteriolar vasoconstriction (mediated by angiotensin II). In this setting, AKI may be precipitated by agents that impair afferent arteriolar dilation (nonsteroidal anti-inflammatory drugs [NSAIDs]) or efferent vasoconstriction (angiotensin-converting enzyme [ACE] inhibitors and angiotensin receptor blockers [ARBs]). Pre-renal AKI is commonly secondary to extracellular fluid volume depletion resulting from gastrointestinal losses (diarrhea, vomiting, prolonged nasogastric drainage), renal losses (diuretics, osmotic diuresis in hyperglycemia), dermal losses (burns, extensive sweating), or possibly sequestration of fluid, so-called third spacing (e.g., acute pancreatitis, muscle trauma). Renal perfusion may be impaired even in the setting of normal or even increased extracellular fluid. For example, renal perfusion may be reduced by a decreased cardiac output (heart failure) or by systemic arterial vasodilation with redistribution of cardiac output to extrarenal vascular beds (e.g., sepsis, liver cirrhosis). The presence of AKI in the setting of severe heart failure has been termed the cardiorenal syndrome and is often exacerbated by the use of ACE inhibitors and diuretics. An unusual cause of pre-renal AKI is the hyperoncotic state. Infusion of large quantities of osmotically active substances such as mannitol, dextran, or protein can increase the glomerular oncotic pressure enough to exceed the glomerular capillary hydrostatic pressure, which stops glomerular filtration, leading to an anuric form of AKI. Pre-renal AKI can be corrected if the extrarenal factors causing the renal hypoperfusion are rapidly reversed. Failure to restore renal blood flow (RBF) during the functional pre-renal stage will ultimately lead to ischemic ATN and tubular cell injury. In any patient presenting with AKI, an obstructive cause must be excluded because prompt intervention can result in improvement or complete recovery of renal function (see Chapter 60). Post-renal forms of AKI are divided into intratubular and extrarenal. Tubular precipitation of insoluble crystals (phosphate, oxalate, uric acid, methotrexate, acyclovir, sulfonamides, indinavir, triamterene) or protein (hemoglobin, myoglobin, paraprotein) can increase intratubular pressure. If sufficiently high, this opposes glomerular filtration pressure and can decrease GFR. Similarly, obstruction of the extrarenal collecting system at any level (renal pelvis, ureters, bladder, or urethra) can lead to post-renal AKI. Obstructive uropathy is common in older men with prostatic disease and in patients with a single kidney or intra-abdominal, particularly pelvic, cancer. Severe ureteral obstruction is also seen with retroperitoneal fibrosis. Most causes of obstructive uropathy are amenable to therapy, and the prognosis is generally good, depending on the underlying disease. Obstructive uropathy is further discussed in Chapter 60. Acute tubular necrosis commonly occurs in high-risk settings, which include vascular and cardiac surgery, severe burns, pancreatitis, sepsis, and chronic liver disease. ATN is responsible for most cases of hospital-acquired AKI and is usually a result of ischemic or nephrotoxic injury. In the intensive care unit, two thirds of cases of AKI are a result of the combination of impaired renal perfusion, sepsis, and nephrotoxic agents.4 The importance of combined injurious mechanisms is also emphasized by experimental data. In animal studies, severe and prolonged hypotension (<50 mm Hg for 2 to 3 hours in the rat) does not cause ATN. Similarly, in animal models, very high doses of single nephrotoxic agents are required to induce AKI. These features may reflect an inherent resistance to tubular injury in animal models, but also illustrate the fact that a single insult alone is rarely sufficient to induce ATN. Fever may exacerbate ATN by increasing the renal tubular metabolic rate, thereby increasing adenosine triphosphate (ATP) consumption. In an experimental model (renal artery occlusion in the rat), renal ischemia for 40 minutes resulted in minimal renal injury at 32° C but marked renal injury at 39.4° C. The typical course of uncomplicated ATN is recovery over 2 to 3 weeks; however, superimposed renal insults often alter this pattern. For example, episodes of hypotension induced by hemodialysis may lead to additional ischemic lesions, potentially prolonging renal functional recovery, and patients with AKI often have multiple comorbidities. The typical features of ATN on renal biopsy include vacuolization and loss of brush border in proximal tubular cells. Sloughing of tubular cells into the lumen leads to cast obstruction, manifested by tubular dilation. Interstitial edema can produce widely spaced tubules, and a mild leukocyte infiltration may be present (see Fig. 69-3). Despite the term acute tubular “necrosis,” frankly necrotic cells are not a common finding on renal biopsy, and histologic evidence of injury frequently involves only 10% to 15% of the tubules despite marked functional impairment. This implies that factors other than just tubular cell injury (such as vasoconstriction and tubular obstruction) are important in the loss of GFR. The tubular damage is usually caused by a combination of ischemic injury resulting in depletion of cellular ATP and direct tubular epithelial cell injury by nephrotoxins. Most accept that the S3 segment of the proximal tubule and the medullary thick ascending limb (mTAL) are particularly vulnerable to hypoxic injury (Fig. 69-4). There are several reasons for this vulnerability.31 The blood flow to the kidney is not uniform, and most of it is directed to the renal cortex, where the cortical tissue partial oxygen pressure (Po2) is 50 to 100 mm Hg, for glomerular filtration. By contrast, the outer medulla and medullary rays are watershed areas receiving their blood supply from vasa recta. Countercurrent oxygen exchange occurs, leading to a progressive fall in Po2 from cortex to medulla, which results in medullary cells living on the “brink of hypoxia” (medullary Po2 as low as 10 to 15 mm Hg). The S3 segments of proximal tubule cells and distal mTALs are thus exposed to borderline chronic oxygen deprivation. The cells of the S3 region and mTAL have high metabolic activity, principally because of sodium reabsorption driven by basolateral membrane Na+-K+-ATPase. Indeed, blocking sodium reabsorption in the mTAL with loop diuretics raises the medullary tissue Po2 from about 15 to 35 mm Hg. Of note, the reduction of GFR in the setting of AKI may be renoprotective by diminishing sodium filtration and hence limiting ATP-dependent sodium reabsorption. Proximal tubular cells have minimal glycolytic machinery and rely almost solely on oxidative phosphorylation for the generation of ATP. In contrast, mTAL cells have a large glycolytic capacity and are more resistant to hypoxic or ischemic insults. Autoregulation normally occurs between systolic blood pressures of 80 and 150 mm Hg; within this range, RBF, glomerular pressures, and GFR are maintained. Below 80 mm Hg, this autoregulation fails, and ischemic injury may result. In certain conditions, such as aging or chronic renal disease, autoregulation is abnormal, and ischemic injury may occur more easily with reductions in perfusion pressure. In addition, experimental studies demonstrate impaired autoregulation in ischemic ATN. In settings of low renal perfusion (e.g., volume depletion, left ventricular failure, edematous states, renal artery stenosis), GFR may be dependent on autoregulation mediated by vasodilatory prostaglandins acting on the afferent arteriole and angiotensin II–mediated efferent arteriolar vasoconstriction to maintain glomerular pressure. Any interference with these mechanisms (e.g., administration of ACE inhibitors or acute inhibition of cyclooxygenase-1 [COX-1] or COX-2 by NSAIDs) may produce a precipitous fall in GFR. In established ATN, RBF is decreased by 30% to 50%. Indeed in AKI, rather than the normal autoregulatory renal vasodilation that occurs in response to decreased perfusion pressure, there is evidence of renal vasoconstriction. A number of vasoconstrictors have been implicated in this response, including angiotensin II, endothelin-1, adenosine, thromboxane A2, prostaglandin H2, leukotrienes C4 and D4, and sympathetic nerve stimulation (Fig. 69-5). Tubuloglomerular feedback (TGF) also contributes to this vasoconstriction. Some of these vascular abnormalities may be mediated by increased cytosolic calcium content in afferent arterioles as a result of ischemia. Disruption of the actin cytoskeleton in vascular smooth muscle cells may also impair autoregulation. The role of TGF (see Chapter 2) in the setting of AKI may be partly beneficial because the resultant decrease in GFR limits sodium delivery to damaged tubules and decreases ATP-dependent tubular reabsorption, protecting against intracellular ATP depletion augmenting renal injury. In this respect, adenosine A1 receptor knockout mice with absent TGF have augmented AKI after ischemia reperfusion. Acute kidney injury is not limited to the tubular cell, and endothelial cell injury occurs partly as a result of acute renal ischemia and oxidant injury.5 Endothelial injury is characterized by cell swelling, upregulation of adhesion molecules (with enhanced leukocyte–endothelial cell interactions), and impaired vasodilation (decreased endothelial nitric oxide synthase and vasodilatory prostaglandins) and may mediate some of the impaired autoregulation and intrarenal vasoconstriction described earlier. Endothelial injury within the peritubular capillaries (vasa recta) may produce congestion in the outer medulla, exacerbating hypoxic injury to the S3 segment of the proximal tubule and the thick ascending loop of Henle. The tubular cell may be injured because of ischemia, with resulting depletion of cellular energy stores (ATP), or from direct cytotoxic injury. Following acute renal ischemia, tubular cell injury may also result from the restoration of RBF (reperfusion injury). Mediators of tubular cell injury include reactive oxygen species (ROSs), intracellular calcium influx, nitric oxide, phospholipase A2, complement, and cell-mediated cytotoxicity. Mitochondrial injury can be caused by ROSs, depletion of antioxidants, and increased intracellular calcium. Disruption of mitochondrial function then exacerbates cellular injury because of disrupted energy metabolism and release of proapoptotic proteins. Autophagy is a mechanism by which cells degrade proteins, and it is a central part of the cellular response to stress and injury. Experimental work has shown that autophagy is important for removal of damaged mitochondria and recovery of tubular epithelial cells from ischemic injury. ROSs may be derived from local sources (including xanthine oxidase and COXs, and secondary to mitochondrial injury) or from infiltrating leukocytes. In models of ischemic ATN, a variety of methods that inhibit ROSs protect against renal injury.6 Hypoxia inducible factor (HIF) and downstream mediators such as heme oxygenase 1 may protect cells against ischemic injury.7 Factors that affect the integrity and function of the renal tubular epithelial cells and contribute to the reduction in GFR include the following (Fig. 69-6): Cell death—Despite the term acute tubular necrosis, only a small percentage of tubular cells undergo cell death, and of these, many actually die by apoptosis rather than necrosis. Indeed, recent studies in animal models have shown amelioration of renal injury with use of caspase inhibitors and p53 inhibitors that decrease apoptosis.8 Although ischemia causes direct renal cytotoxicity, tissue inflammation during reperfusion also contributes to renal injury and may cause some of the systemic effects of AKI. Components of both the innate and the adaptive immune systems contribute to the pathogenesis of ATN.9 The innate immune system is activated by cellular injury and certain pattern recognition molecules. Toll-like receptor 2 (TLR2) and TLR4 are upregulated within the kidney after ischemia, are activated by molecules released from injured cells, and induce renal epithelial cells to produce chemokines. The complement system is also activated within the tubulointerstitium after ischemia and reperfusion, predominantly by the alternative pathway. It can directly induce nearby epithelial cells to produce proinflammatory cytokines (such as tumor necrosis factor α [TNF-α], interleukin [IL]–6, IL-1β) and chemokines (such as monocyte chemoattractant protein 1[MCP-1], IL-8, regulated upon activation normal T cell expressed and presumably secreted [RANTES]) that promote the infiltration of the kidney by leukocytes and are also directly vasoactive. A network of dendritic cells extends throughout the renal tubulointerstitium; these cells help shape the inflammatory response within the kidney after ischemia and reperfusion, likely through their interactions with other inflammatory cell types. Neutrophils and mononuclear cells are seen in peritubular capillaries on renal biopsy. Experimental studies have shown that neutrophil activation and the release of proteases and ROSs can exacerbate injury. By contrast, neutrophil depletion with antibody, or inhibiting neutrophil adhesion molecules (ICAM-1) with antibody or antisense oligonucleotides, ameliorates injury in ischemic ATN. ICAM-1 knockout mice are similarly protected. Monocytes infiltrate the reperfused kidney and differentiate into the M1, or inflammatory, type. M1 macrophages seem to exacerbate renal injury after ischemia, but studies have demonstrated that macrophages may later convert to the M2 “anti-inflammatory” type and promote repair of the kidney. A number of studies have also demonstrated that cells of the adaptive immune system, including T and B lymphocytes, contribute to renal injury in models of ATN. Experimental ischemic reperfusion injury may be ameliorated by T cell or B cell deficiency. It is not known whether these responses are antigen specific. Furthermore, some B and T cell subsets, such as T regulatory cells, help limit renal injury. We should also recognize that AKI may have systemic effects on other organ systems.10 The injured kidney may prime and activate leukocytes, with the production of proinflammatory cytokines, which can mediate remote organ injury (Fig. 69-7). The lungs may be particularly vulnerable from the combined effects of volume overload, increased vascular permeability, and the proinflammatory environment. These distant organ effects may partly account for the increased mortality in patients with AKI (Chapter 72).
Pathophysiology and Etiology of Acute Kidney Injury
Definition
Etiologic Overview
Pathophysiology and Etiology of Pre-Renal Acute Kidney Injury
Pathophysiology and Etiology of Post-Renal Acute Kidney Injury
Pathophysiology of Acute Tubular Necrosis
Histology
Tubular Injury in Acute Tubular Necrosis
Blood Supply
High Tubular Energy Requirements
Glycolytic Ability of Tubular Cells
Hemodynamic Factors in the Development of Acute Tubular Necrosis
Impaired Renal Autoregulation
Intrarenal Vasoconstriction
Tubuloglomerular Feedback
Endothelial Cell Injury and the Development of Acute Tubular Necrosis
Tubular Epithelial Cell Injury and the Development of Acute Tubular Necrosis
Inflammatory Factors in the Development of Acute Tubular Necrosis
Recovery Phase
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pathophysiology and Etiology of Acute Kidney Injury
Chapter 69