 FIGURE 3-1 ▪ Adrenal cortex of a 16-week fetus demonstrating the broad inner zone of eosinophilic cells (provisional zone or fetal cortex) and the thin outer zone that will form the adult cortex. |
Table 3-1 ▪ AVERAGE COMBINED WEIGHTS OF ADRENAL GLAND FROM SERIES OF AUTOPSY PATIENTS* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
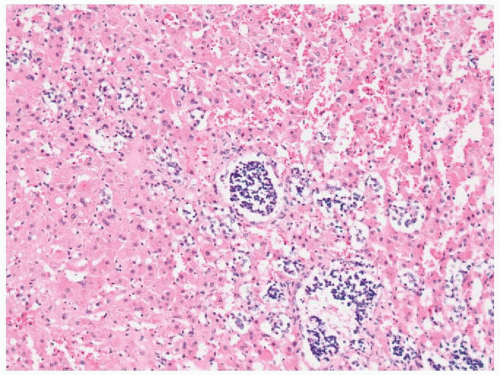 FIGURE 3-2 ▪ Groups of primitive sympathetic cells forming neuroblastic nodules within the fetal cortex in the adrenal gland of a 16-week fetus (same specimen as Fig. 3-1). |
during the process of recovery. The remainder of the cortex is composed of the reticularis, which is capable of synthesis of both glucocorticoids and sex steroids. The cells of the reticularis have eosinophilic cytoplasm, scanty lipid vacuoles, and prominent deposits of lipochrome pigment, which are responsible for the brown color of the reticularis. Ultrastructurally, the cells of the glomerulosa contain lamelliform mitochondria while tubulovesicular mitochondria predominate in the fasciculata and reticularis. The intermediate filaments of cortical cells include vimentin and variable amounts of low-molecular-weight cytokeratins.12 Cortical cells are also positive for calretinin, inhibin A, melan-A, and synaptophysin.13, 14, 15, 16
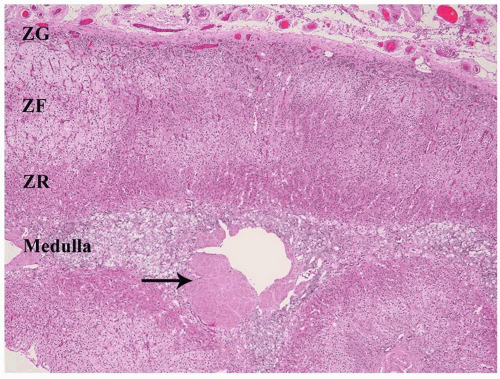 FIGURE 3-3 ▪ Histology of the normal adrenal gland. The three cortical zones (ZG, zona glomerulosa; ZF, zona fasciculata; ZR, zona reticularis), and medulla (MED) are demonstrated. There is slight nodularity of the cortex as occurs in normal adults. Also demonstrated is the eccentric smooth muscle wall of the central vein (arrow). |
(CRH), a 41-amino-acid polypeptide, reaches the anterior pituitary gland via the hypophyseal portal system, where it stimulates the release of ACTH.20
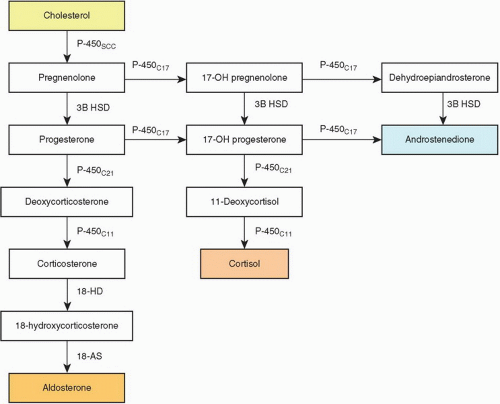 FIGURE 3-4 ▪ Biosynthesis of adrenal steroid hormones. 18-AS, 18-aldehyde synthetase; 3B HSD, 3-β hydroxysteroid dehydrogenase; 18 HD, 18 hydroxylase. |
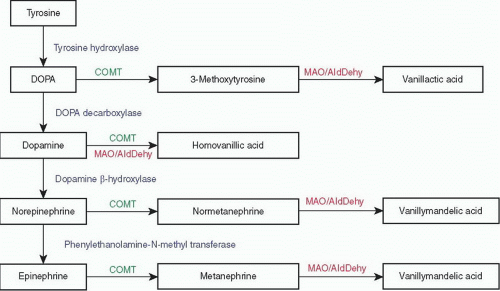 FIGURE 3-5 ▪ Catecholamine biosynthesis and metabolism. COMT, catecholamine-O-methyl transferase; DOPA, 3,4-dihydroxyphenylalanine; MAO/AldDehy, monoamine oxidase and aldehyde dehydrogenase. |
few examples, particularly those in the region of the celiac ganglion, may also contain medullary tissue.37 Based on autopsy studies accessory adrenal tissue is most frequently found in the retroperitoneal space (32%) along the celiac axis, the upper pole of the kidney just beneath the renal capsule (0.1% to 6%), the broad ligament (23%), adnexa of the testes (7.5%), and along the course of the spermatic cord (3.8% to 9.3%).11 Less common sites include the pancreas, spleen, liver, lung placenta, and brain. In surgical pathology series, the most commonly encountered site is in inguinal hernia sacs. One percent of hernia sacs have been reported to contain accessory adrenal tissue.38 Accessory adrenals may undergo hyperplasia in response to increased levels of ACTH and may serve as the site of origin of cortical neoplasms.23,39,40 True heterotopic adrenal glands may be fused with the liver or kidney and are typically surrounded by a common connective tissue capsule.41,42 In such cases, a concern for a metastatic renal cell carcinoma (RCC) or other clear-cell tumors may arise.
are predisposed to the development of a variety of malignant tumors including Wilms tumor, adrenal cortical carcinoma, neuroblastoma (NB), hepatoblastoma, and pancreaticoblastoma. However, the hyperplastic cortical cells in this syndrome do not represent a preneoplastic lesion.43,50 Adrenal cortical adenomas and ganglioneuromas have also been reported in affected patients.49 Autosomal dominant inheritance is well established in Beckwith-Wiedemann syndrome, but approximately 85% of cases are actually sporadic. The molecular basis of this syndrome is complex and involves downregulation of imprinted genes within the chromosome 11p15 region (Table 3-2). Other syndromes that predispose to adrenal tumors are discussed in subsequent sections.
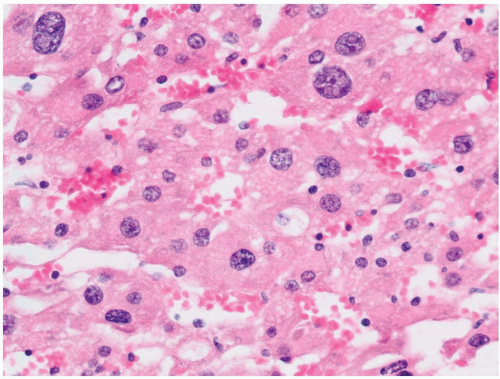 FIGURE 3-6 ▪ Focal cytomegaly of cells of the fetal cortex from a newborn. |
Table 3-2 ▪ SYNDROMES ASSOCIATED WITH ADRENALTUMORS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
is tragically rapid and the child is often in a vegetative state within 1 to 2 years. According to Moser et al.53 there are seven phenotypes that include the childhood cerebral form, adrenomyeloneuropathy, adult cerebral, adolescent, adrenal insufficiency without neurologic disease, asymptomatic, and heterozygotes. Clinical manifestations can, therefore, vary widely even within the same family.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
a common cause in parts of the world where tuberculosis is endemic. In contrast to the shrunken appearance of the adrenal glands in idiopathic Addison disease, the glands in mycobacterial infection are typically enlarged and necrotic. Infection with Mycobacterium avium intracellulare is typically associated with the presence of confluent masses of histiocytes containing acid-fast organisms.
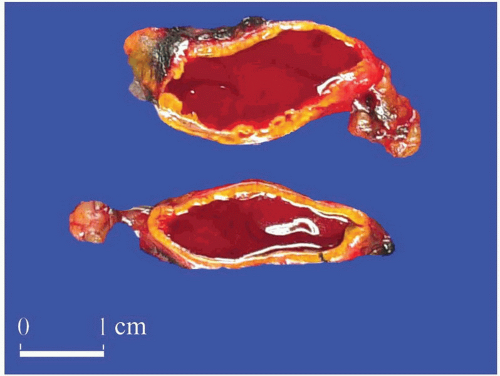 FIGURE 3-7 ▪ Adrenal gland hemorrhage in a patient with cholangiocarcinoma. There was no evidence of metastatic disease. |
configurations of the glands are retained. Typically, the cortex is bright yellow owing to lipid accumulation in the cortical cells, the capsule is fibrotic, and the medulla is unaffected. The zona glomerulosa is usually of normal thickness in these cases.
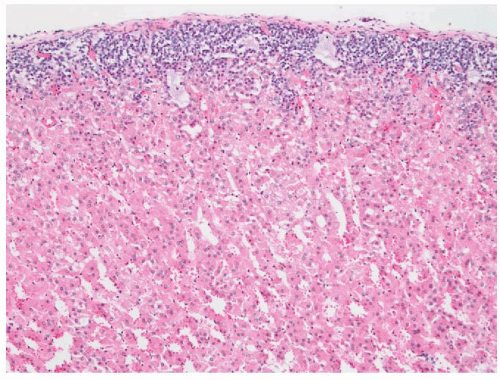
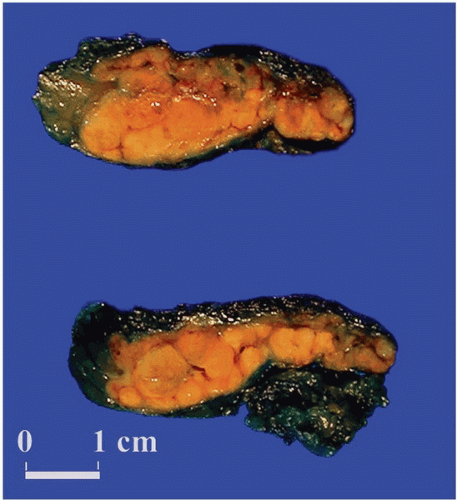 FIGURE 3-8 ▪ Diffuse and nodular adrenocortical hyperplasia. Adrenal gland from a patient with pituitary-dependent Cushing syndrome (Courtesy Dr. A. Mc Nicol, Glasgow, Scotland, UK.). |
a compact or lipid-depleted appearance.82 Foci of nuclear enlargement and hyperchromasia of reticularis cells may be noted, and these features may be particularly striking adjacent to metastatic foci in the glands.23 Both bronchial carcinoids and small-cell bronchogenic carcinomas may also produce CRH.
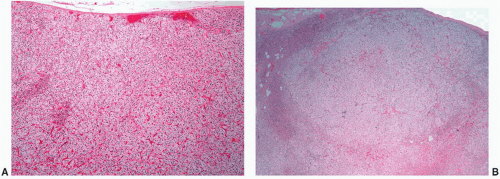 FIGURE 3-9 ▪ Diffuse and nodular adrenocortical hyperplasia. Histologic section of diffuse (A) and nodular hyperplasia (B) from a patient with Cushing syndrome. There is predominantly hyperplasia of the vacuolated fasciculata. |
Carney complex (CNC), which includes cardiac myxomas, spotty pigmentation, neurofibromatosis, testicular Leydig or Sertoli cell tumors, mammary myxoid fibroadenomas, and cerebral hemangiomas.89,91 The Carney complex may occur sporadically or may be inherited as an autosomal dominant trait. The gene encoding the protein kinase A type Iα regulatory subunit PRKAR1A has been mapped to 17q22-q24 and loss of heterozygosity (LOH) studies from patients with CNC have revealed mutations in this gene in approximately 50% of affected individuals. No mutations have been found on 2p16. Studies of sporadic and isolated cases of (CNC) have also revealed inactivating mutations of PRKAR1A. The wild-type alleles could be inactivated by somatic mutations consistent with the hypothesis that the gene belongs to the tumor suppressor class.92,93 Some studies have suggested that PPNAD may have an autoimmune etiology.94
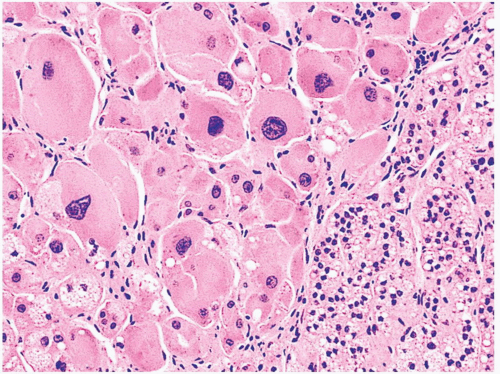 FIGURE 3-10 ▪ Primary pigmented nodular adrenocortical disease (PPNAD). The nodule is composed of large cells with hyperchromatic nuclei. In this photomicrograph pigment is absent. |
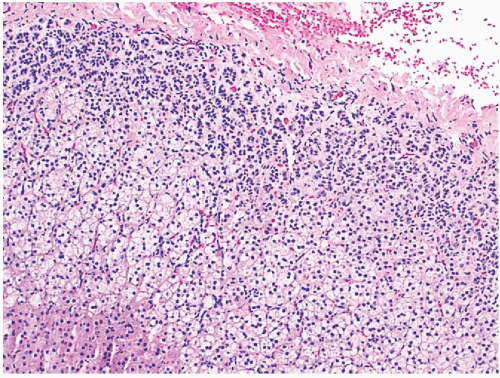 FIGURE 3-11 ▪ Hyperplasia of the zona glomerulosa in a patient with primary hyperaldosteronism. |
cords, or alveolar arrangements of vacuolated (clear) cells that most closely resemble those of the normal fasciculata (Fig. 3-12B). Variable numbers of compact-type cells are also present (Fig. 3-12C). Black adenomas are composed exclusively of lipochrome-rich compact cells.6 The nuclear/cytoplasmic ratio is generally low, although a few single cells and small groups of cells may have enlarged and hyperchromatic nuclei. Typically, the nuclei are vesicular with small, distinct nucleoli and little or no mitotic activity. In fine-needle aspiration biopsy specimens, the cells are round to polyhedral with round nuclei and foamy cytoplasm. Numerous naked nuclei in a background of granular to foamy material may be prominent.
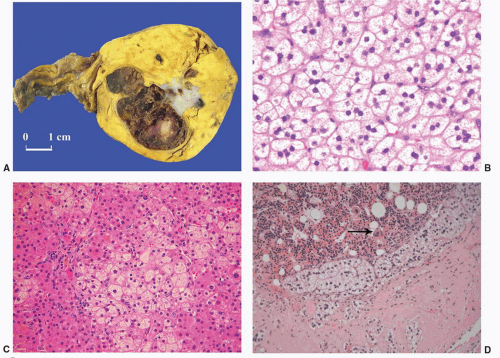 FIGURE 3-12 ▪ Nonfunctional adrenocortical adenoma. The cut surface is bright yellow with central hemorrhage and fibrosis (A) (Courtesy of Dr. A. Matoso, Providence, RI). The tumor is composed predominantly of clear cells that resemble normal cells of the fasciculata (B), but compact cells are also present (C). Myelolipomatous change was present in a section from the central hemorrhagic area (D, arrow megakaryocyte). |
be prominent. Foci of myelolipomatous change or calcification may be seen, particularly in larger adenomas.23 On ultrastructural examination, adenoma cells most closely resemble the cells of the normal fasciculata or reticularis.6
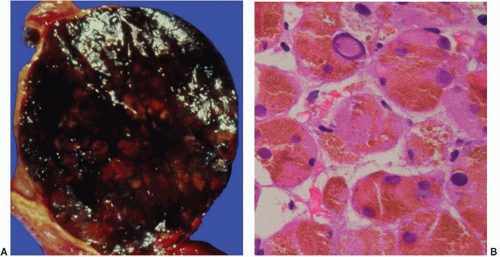 FIGURE 3-13 ▪ Pigmented (black) adenoma associated with Cushing syndrome (A). The tumor is composed predominantly of compact cells containing abundant lipochrome pigment (B). |
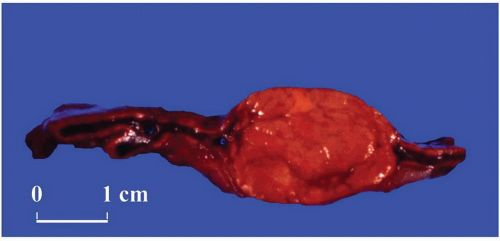 FIGURE 3-14 ▪ Adrenocortical adenoma associated with Conn syndrome. This tumor has a yellow-brown color or orange cut surface rather than a bright yellow appearance that is more characteristic. |
weighed up to 500 g.9,23,43 Similar to tumors associated with glucocorticoid overproduction, virilizing adenomas are sharply circumscribed or encapsulated; however, they tend to be red-brown rather than yellow on cross section23 (Fig 3.16A). Smaller tumors have an alveolar pattern of growth, whereas larger tumors tend to have more solid or diffuse growth patterns. Although most tumor cells have a low nuclear/cytoplasmic ratio, single cells and small-cell groups may exhibit considerable nuclear enlargement and hyperchromasia. The cytoplasm is usually eosinophilic and granular (Fig. 3.16B). Rare virilizing tumors contain Reinke crystalloids and have been termed Leydig cell adenomas similar to their testicular counterparts.105,106 On ultrastructural examination, the mitochondria are of the tubulolamellar type. Sex steroid-producing adenomas are not associated with atrophy of the adjacent cortex or the contralateral adrenal gland.
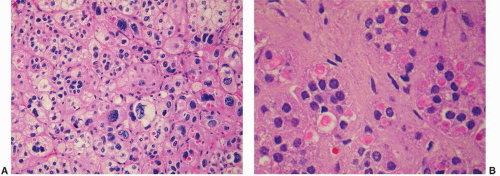 FIGURE 3-15 ▪ Adrenocortical adenoma associated with Conn syndrome. The tumor is composed of an admixture of fasciculata- and glomerulosa-type cells. Cells with enlarged, hyperchromatic nuclei are evident (A). Spironolactone bodies that have a typical lamellar appearance are prominent in this section (B). |
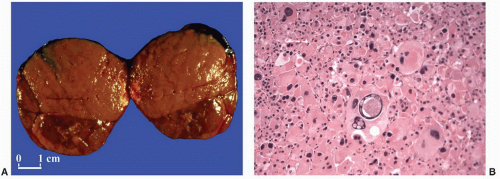 FIGURE 3-16 ▪ Adrenocortical adenoma associated with virilizing syndrome. This tumor from a 2-year-old girl has a brown cut surface (A). The neoplastic cells are eosinophilic and granular with focal nuclear enlargement and hyperchromasia (B). Nuclear pseudoinclusions are also present. |
tumors were composed of 50% to 90% oncocytic cells. Subsequently, Duregon et al.113b classified oncocytic adrenocortical tumors as being pure, mixed, having focal oncocytic features, or being conventional adrenocortical tumors when the oncocytic cells constituted >90%, 50% to 90%, 10% to 49%, and <10% of the tumor cells, respectively (Box 3-2). Because of variations in sampling with fine needle or core biopsies it is appropriate to render a diagnosis of “adrenocortical neoplasm with oncocytic features” in such samples.
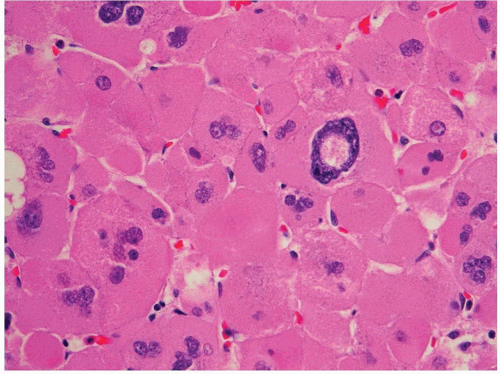 FIGURE 3-17 ▪ Adrenocortical oncocytoma. The cells contain densely granular eosinophilic cytoplasm and pleomorphic nuclei with a pseudoinclusion. |
| ||||||||||||||||||||||||||||||||||||
present. A significant proportion of cortical carcinomas (up to 75% in some series) may be unassociated with syndromes of hormone overproduction.120 In some instances, patients may show signs of hypoglycemia due to the production of IGFs by the tumor or hypercalcemia due to the production of parathyroid hormone-related peptide.
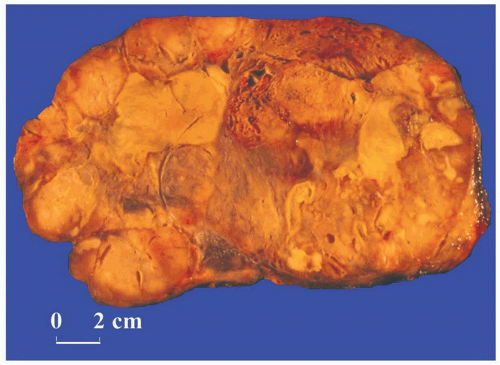 FIGURE 3-18 ▪ Adrenocortical carcinoma. The cut surface of this large tumor shows prominent necrosis. |
of these patterns.23,37,43 Many tumors are composed of widened trabeculae separated by endothelium-lined sinusoidal channels (Fig. 3-20A) in contrast to the thin cell cords characteristic of adenomas. Necrosis, particularly in large tumors, may be extensive (Fig. 3-21A). Foci of myxoid change, pseudoglandular patterns, and spindle-cell growth may be prominent in some cases.126 Depending on their lipid content, the cytoplasm may vary from vacuolated to eosinophilic. Some tumors may have eosinophilic globular inclusions resembling those seen in pheochromocytomas. Rare cases may exhibit adenosquamous differentiation.127 There may be considerable variation in the appearance of the nuclei. In some instances, they may appear relatively small and uniform, while in others they may exhibit marked pleomorphism (Fig. 3-20B), coarse chromatin, and multiple enlarged nucleoli. Mitotic activity, including atypical forms (Fig. 3-20C), is often prominent. Nuclear pseudoinclusions may be particularly striking in some cortical carcinomas.
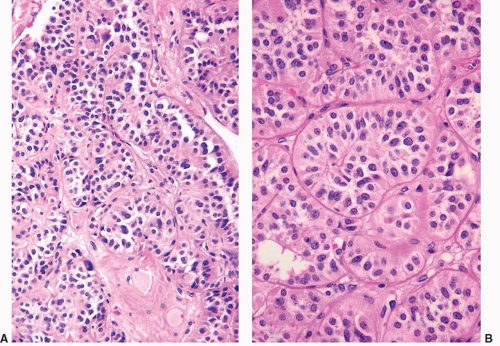 FIGURE 3-19 ▪ Adrenocortical carcinoma. The tumor has an alveolar pattern of growth (A, low power; B, high power). |
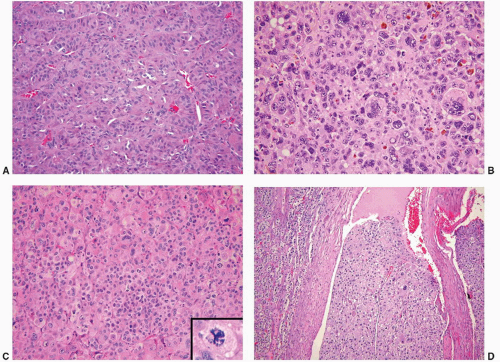 FIGURE 3-20 ▪ Adrenocortical carcinoma. Trabecular growth pattern (A), nuclear pleomorphism (B), prominent mitotic activity (C, inset atypical mitotic figure), and vascular space invasion (D) are demonstrated. |
Fischler et al.,131 the sarcomatous component had features of rhabdomyosarcoma and stained positively for musclespecific actin and desmin. Although this tumor was associated with virilization, the case reported by Decorato et al.132 was nonfunctional. Recently, Thway et al.132a reported an example of oncocytic adrenocortical carcinosarcoma in a 45-year-old man with pleomorphic rhabdomyosarcoma that metastasized to the mesentery, hilar lymph nodes, lungs, and brain over the 11-month terminal course from diagnosis.
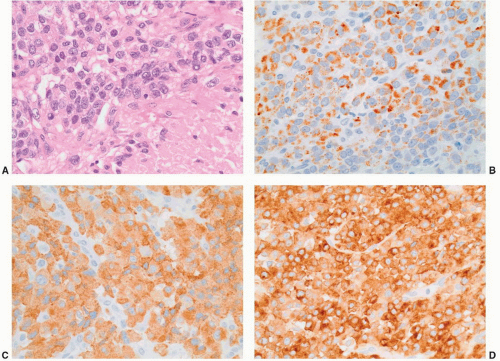 FIGURE 3-21 ▪ Adrenocortical carcinoma. There was extensive necrosis of this tumor and viable tumor cells have high nuclear to cytoplasmic ratio (A, hematoxylin-eosin section; necrosis in right lower aspect). Neoplastic cells stain positively with antibodies to melan-A (B), inhibin (C), and synaptophysin (D). |
cells. Renshaw and Granter15 have demonstrated that inhibin A and A103 are both useful for the identification of adrenal cortical neoplasms and that A103 is marginally more specific and inhibin A slightly more sensitive. Calretinin is also expressed in adrenal cortical neoplasms and is a useful adjunct in cases where stains for inhibin A are negative. Jorda et al.13 demonstrated that almost 75% of cortical neoplasms were positive for inhibin A; however, when calretinin was added, the numbers of tumors staining positively for the two markers increased to 94% (31/33 cases).
Table 3-3 ▪ DIFFERENTIAL DIAGNOSIS OF ADRENOCORTICAL CARCINOMA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||









