Nonneoplastic Lesions of the Testis
FERRAN ALGABA
MUKUL K. DIVATIA
ALBERTO G. AYALA
JAE Y. RO
TESTICULAR EMBRYOLOGY
Genetic Mechanisms Regulating Testicular Development and Sex Determination
Although multiple genes play a role in testicular differentiation, the two genes of prime importance are NR5A1 and WT-1 (Wilms tumor gene). NR5A1 is located on chromosome 9q33.3 and comprises seven exons with a gene product known as SF-1 (steroidogenic factor 1). SF-1 is initially recovered from the Sertoli cells of the sex cords, but it is localized to the Leydig cells during later developmental stages.1 It enhances the expression of anti-müllerian hormone (AMH) and plays a role in regulation of the AMH gene. A female phenotype is found in 46XY subjects with a heterozygous deletion of NR5A1 along with other features including adrenal failure in the first months after birth, persistence of normal müllerian structures and maldeveloped gonads comprising poorly differentiated tubules with abundant connective tissue stroma. The neonatal phenotype is not a reliable predictor of virilization at puberty. Male gender assignment in poorly virilized cases at birth may allow spontaneous puberty without signs of hypogonadotropic hypogonadism, and possibly fertility. Patients with SF-1 mutations are at increased risk for malignant germ cell tumors. Early orchidopexy and germ cell tumor screening are mandated in the presence of preserved gonads. In cases where premalignant and/or malignant changes are identified, gonadectomy with or without possible irradiation constitutes the modality of treatment.2 Adrenal failure is the sole presenting feature in patients with 46XX since SF-1 does not influence ovarian development.3 These disorders aid in highlighting the significant position of NR5A1 expressed in the primitive urogenital ridge, which differentiates into the gonads and adrenal glands.
WT1 is located on chromosome 11p13 and contains 10 exons with alternative splicing sites in introns 5 and 9. The splicing of intron 9 can result in KTS+ (three amino acids viz. lysine, threonine, and serine) or KTS- isoforms. Normal gene expression mandates a proper balance of both isoforms. The WT1 gene is predominantly expressed in the kidneys and gonads. It is responsible for stromal-epithelial transition and inhibition of genes encoding proliferative factors for epithelial differentiation and simultaneous activation of genes enhancing the process. A host of phenotypic alterations are observed with WT1 gene anomalies.4 Loss of the KTS+ isoform gives rise to Frasier syndrome characterized by 46XY gonadal dysgenesis, absence of Wilms tumor, and renal disease of late-onset type.5 Missense heterozygous mutations are manifested as Denys-Drash syndrome with partial or complete 46XY gonadal dysgenesis, Wilms tumor, and earlyonset renal disease with diffuse mesangial sclerosis.6 WT1 deletions are linked to an increased propensity to develop Wilms tumor and variable genitourinary system manifestations. Other genes identified in the formation of kidneys and gonads are LIM1 and FGF-9 (fibroblast growth factor 9).
The signal for gonadal differentiation is initiated by the SRY gene on the sex-determining region of chromosome Y (Yp11.3) otherwise known as testis determining factor gene.7 It is this gene that is responsible for production of the AMH, differentiation of Sertoli cell precursors and germ cells, and downstream gene regulation.8 The pathway of testicular differentiation is complex and involves activation and inhibition of both autosomal and sex chromosomal genes. The SRY gene has been demonstrated in the nuclei of germ cells and Sertoli cells. It contains a single exon encoding a 204-amino acid protein of the central part that is responsible for encoding a DNA-binding domain referred to as high mobility group (HMG). SRY also regulates steroid hormonal expression and interacts with the AMH promoter gene.9 Mutations of SRY give rise to pure gonadal dysgenesis (Swyer syndrome) or true hermaphroditism. Although all affected cases possess male external genitalia and testes, they have no müllerian structures and azoospermia. The karyotype of patients with the male phenotype without the Y chromosome is 46XX SRY+ in 80% cases and 46XX SRY- in 20% cases. SOX9 duplication may also be present in some instances.10
Several other genes encode associated transcription factors and play a role in gonadal differentiation including DAX-1, SOX-8, SOX-9, LHX-9, LIM-1, and DMRT-1. DAX-1 (dosage-sensitive sex reversal) gene is situated on X chromosome and is part of the pathway for development of ovaries, testes, and adrenal glands. It is inhibited by SRY during testicular differentiation and activated during ovarian differentiation. DAX mutations are associated with decreased levels of gene expression causing nondevelopment of the adrenal cortex and hypogonadotrophic hypogonadism with normal testicular development.11 Human DAX1 duplications result in dosage-sensitive sex reversal (DSS) subsequent to which individuals with a chromosomal XY pattern can develop as females due to gonadal dysgenesis. The exact mechanism of DSS-adrenal hypoplasia congenita on X, gene 1 (DAX1) action in the fetal testis is albeit unknown. It has been demonstrated that in fetal testes from XY Dax1-overexpressing transgenic mice, the expression of the key testis-promoting gene sex-determining region on Y (SRY)-box-9 (Sox9) is reduced. Also, in XY Sox9 heterozygotes, in which testis development is usually normal, Dax1 overexpression results in ovotestes, thereby indicating a DAX1-SOX9 antagonism. The ovarian portion of the XY ovotestes in a recent study was characterized by expression of the granulosa cell marker, forkhead box-L2, with complete loss of the Sertoli cell markers, SOX9 and AMH, and the Leydig cell marker CYP17A1. However, the expression of SRY and SF-1, two key transcriptional regulators of Sox9, was retained in the ovarian portion of the XY ovotestes. Dax1 overexpression reduced activation of TES, the testis enhancer of Sox9, indicating that DAX1 might repress Sox9 expression via TES in reporter mice. Increasing levels of DAX1 antagonized SF-1-, SF-1/SRY-, and SF-1/SOX9-mediated activation of TES in cultured cells, as a result of reduced binding of SF-1 to TES, thus providing a possible mechanism for DSS.12
SOX-8 and SOX-9 (SRYY box 8 and 9) are linked to autosomal genes. SOX9 is located on chromosome 17q24. 3q25.1 and is expressed after SRY expression in pre-Sertoli cells.13 The protein encoded by this gene recognizes the sequence CCTTGAG along with other members of the HMG-box class DNA-binding proteins. It acts during chondrocyte differentiation and, with SF-1, regulates transcription of the AMH gene. Functional allelic losses lead to the skeletal malformation syndrome (campomelic dysplasia), frequently with 46XY constitution with female phenotype.13 Duplication of SOX-9 results in 46XX patients with male phenotype.14 SOX-8 is another gene involved in AMH regulation and interacts with SF-1 through protein-protein interactions. It has been demonstrated experimentally that SOX-9 dysfunction leads to SOX-8 expression as a replacement through a feedback process.15
Deletions in chromosome 9p16 and 10q17 result in expression of a female phenotype in 46XY genotype cases. Deletions of chromosome 9p are also associated with hydronephrosis, facial malformations, and delayed development. In 46XY females, deletions of two genes (DMRT1 and DMRT2) are located on chromosome 9p24.3. Chromosome 10q deletions are associated with genital malformations, mental retardation, and other systemic manifestations.
Hormonal Control
Multiple hormones are involved in the development of the male genital system at various stages, including AMH, testosterone, dihydrotestosterone (DHT) and the pituitary gland hormones, follicle-stimulating hormone (FSH), and the luteinizing hormone (LH).
AMH (also known as müllerian inhibitory substance, MIS) is a glycoprotein consisting of two identical 72-kDa subunits linked by disulfide bonds that is secreted by Sertoli cells in males and granulosa cells in females.18 Its expression is regulated by SF-1, which is a transcriptional regulator of many steroid genes.19 AMH is a member of the TGF-β family encoded by a 2.75-kb gene situated on 19p13.2. The amount of AMH secreted is inversely proportional to degree of Sertoli cell maturation. It can be detected during the 8th to 9th week of gestation, and its concentration rises in the second trimester and decreases significantly in the third trimester.20 Although its level rises upon birth and AMH is detectable during childhood, its levels decline to undetectable with the onset of puberty when it is negatively regulated by androgens.21
The target sites of AMH include the genital tract, testis, and surrounding structures with AMH causing involution of the ipsilateral müllerian duct beginning from the caudal pole with rapid progression upward. It regulates SRY expression being expressed around the same time frame. The tunica albuginea forms through mesenchymal insertion between primitive sex cords and coelomic epithelium and its development is promoted by AMH.22 This hormone presents a barrier to spermatogonia entering meiosis.23 Of note is the role played by AMH in the development of fetal lungs.24
Testosterone synthesis commences during the 8th week of gestation and is synthesized by Leydig cells, which appear during the 8th week of gestation and constitute approximately 50% of the testicular volume by the 16th week.25 However, the secretion of testosterone is regulated by hCG and LH levels. hCG levels are at their peak during the 11th to 18th weeks and fall to significantly lower levels after this duration. This period of hCG-dependent testosterone secretion is crucial in terms of genital differentiation. Wolffian duct differentiation occurs when testosterone is secreted by the testis on each side and leads to differentiation of epididymis, ductus deferens, and seminal vesicle. Defects in androgen synthesis are evidenced as cryptorchidism and incomplete masculinization.
The enzyme 5α-reductase acts on testosterone to produce DHT, which in turn is responsible for differentiation of the prostate along with external genitalia, male urethra, scrotum, and penis. The midline fusion of labioscrotal folds (day 70) to form the scrotum with the midline raphe is induced by DHT.
The penile urethra is formed by fusion of the urethral folds (day 74), and the genital tubercle subsequently enlarges to form the glans penis. The terminal urethra develops from an invagination of the glans tip. The prostate, urinary bladder, and prostatic urethra are formed from the urogenital sinus.26
The penile urethra is formed by fusion of the urethral folds (day 74), and the genital tubercle subsequently enlarges to form the glans penis. The terminal urethra develops from an invagination of the glans tip. The prostate, urinary bladder, and prostatic urethra are formed from the urogenital sinus.26
The roles played by FSH and LH gain importance toward the last weeks of gestation. LH levels start rising in fetal circulation during the 10th week and peak by the 18th week after which they decline gradually until birth. LH regulates fetal androgen production in the second and third trimesters. FSH is responsible for Sertoli cell mitogenic activity, which peaks at the time of delivery.27
NORMAL TESTICULAR STRUCTURE
Fetal Testis
In their initial stages, germ cells acquire diverse evolutionary morphologic appearances. Three types of germ cells have been identified, and some authors28 have proposed that these should be known as gonocytes (OCT4 positive, c-kit positive), intermediate germ cells (OCT4 low expression/negative, c-kit negative), and prespermatogonia (OCT4 negative, c-kit negative). In the first trimester, most germ cells have a gonocyte phenotype; however, from the 18th week of gestation, prespermatogonia are the most abundant cell type (Fig. 11-1). These data provide evidence for the functional differentiation of human testicular germ cells during the second trimester of pregnancy and argue against these germ cells being considered a homogeneous population.
Prepubertal Testis
From birth until puberty, the testicles develop continuously, but some phenomena permit this period to be subdivided into the following three phases:
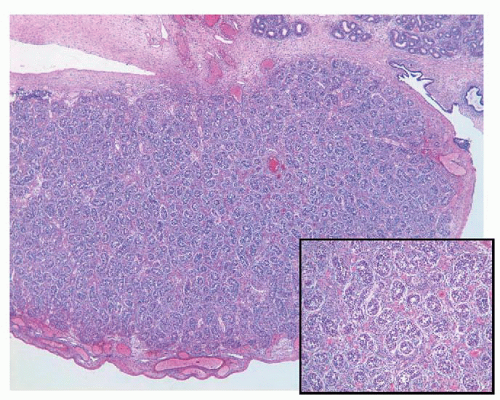 FIGURE 11-1 ▪ Fetal testis. Solid seminiferous tubules with prespermatogonia and interstitial Leydig cells; 24th week of gestation (Inset: Higher magnification). |
Testicle Development in Newborns and Perinatal Period
Testicular development in the newborn is characterized by solid tubules with Sertoli cells and gonocytes (centrally located). At 6 months after birth, there are no gonocytes because they have been transformed into spermatogonia by the testosterone from the Leydig cells.29 The morphologic features of Leydig cells at this stage are similar to those in adults, without Reinke crystalloids.
Testicle Development in Infants
After the changes that occur in the testicle during the first 6 months following birth, the testicle remains at rest until age 3 years. After this time, margination of the cells can be observed, which provides a pseudoluminal appearance of the tubules; in addition, some meiosis can be seen, which rapidly stops in what appears to be cellular rests. The tubular cells are typically composed of Sertoli cells, undifferentiated cells, and some spermatogonia (Fig. 11-2). The Leydig cells have involuted and have decreased in number in this phase, with minimal levels of testosterone.
Testicle Development in Boys
From age 9 years onward and especially between ages 13 and 15 years, the interstitial mesenchymal cells are definitively transformed into adult Leydig cells that produce testosterone under the action of LH; in addition, LH stimulates the development of the germinal cells, growth of the tubule, and appearance of the central lumen.29
Pubertal and Adult Testis
When the testicle is totally developed, it has a supporting structure—the albuginea—that surrounds the testis like an external capsule. Between the thickened mediastinum testis
area and the surface of the organ are fibrous septa that project toward the interior and divide the testicular parenchyma into about 250 segments or lobules. The albuginea has three layers; from outside to inside they include the external mesothelial layer (tunica vaginalis testicular); the medial, relatively acellular layer with fibroblasts, myocytes, and nerve fibers (tunica albuginea); and the internal vascular layer (tunica vasculosa).30
area and the surface of the organ are fibrous septa that project toward the interior and divide the testicular parenchyma into about 250 segments or lobules. The albuginea has three layers; from outside to inside they include the external mesothelial layer (tunica vaginalis testicular); the medial, relatively acellular layer with fibroblasts, myocytes, and nerve fibers (tunica albuginea); and the internal vascular layer (tunica vasculosa).30
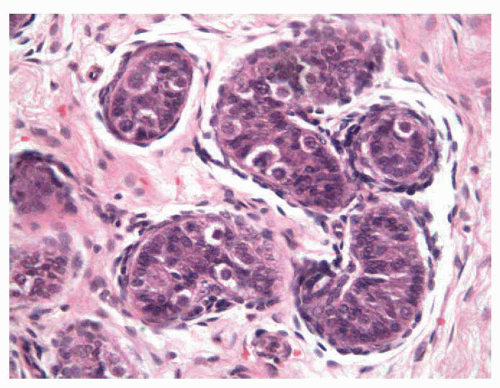 FIGURE 11-2 ▪ Infantile testis. Small solid seminiferous tubules. Immature Sertoli cells with undifferentiated cells and some spermatogonias (cells with halo). |
Table 11-1 ▪ AVERAGE NUMBER OF CELLS PER TUBULE FOR VARIOUS CELLULAR SUBTYPES, PERTRANSVERSE TUBULAR SECTION | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The seminiferous tubules occupy 70% to 75% of the testicular volume. Each seminiferous tubule has a closed-loop structure with intercommunication between the arms of the loop. The size varies according to the section angle. In the cross sections of the tubules in an adult, the average diameter is about 180 μm. Each seminiferous tubule is composed of a tubular wall, Sertoli cells, and germ cells (Table 11-1).
The tubular wall comprises five layers; they include the basement membrane (periodic acid-Schiff positive), the internal acellular layer, the internal myofibroblastic layer, the external acellular layer, and the external fibroblastic layer from the inner to the outer aspect. The innermost strata have contraction functions, apparently useful for intratubular motility, and thus mobilize the spermatozoids toward the rete testis or network of canals at the termination of the straight seminiferous tubules in the mediastinum testis.31 The elastic fibers appear in puberty, are located more externally, and may be lacking in dysgenetic testicles.32
The Sertoli cells (10.2 ± 2 per tubule) have abundant cytoplasm with triangular nuclei and prominent nucleoli that extend from the basal membrane to the tubular lumen. Sertoli cells express vimentin and have the greatest metabolic activity of all cells in the entire tubule since they induce the process of differentiation and the maturation of spermatocytes, regulate the maturation of the Leydig cells, secrete inhibin, and produce tubular fluid.33 These cells have strong desmosomal junctions in their lower portions and weak ones in their upper portions.34 Sertoli cells synthesize and secrete a large variety of factors: proteins, cytokines, growth factors, opioids, steroids, prostaglandins that explain the presence of endoplasmic reticulum smooth and rough type and a prominent Golgi apparatus.
The germ cells progressively mature from spermatogonia to their final mature forms within 70 to 74 days. From the morphologic point of view, 13 distinct types of germ cells can be identified, but at the typical light microscopic level, only the following principal levels of maturation are recognized (Fig. 11-3):
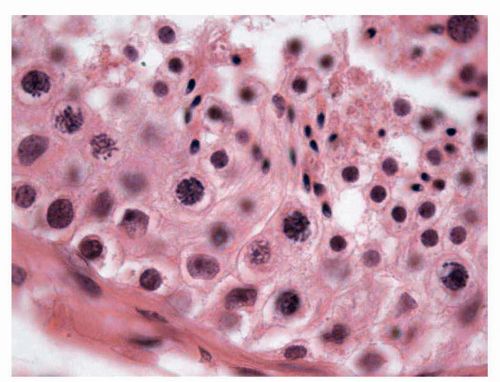 FIGURE 11-3 ▪ Adult testis. Mature Sertoli cells (with triangular nuclei and prominent nucleoli). Normal spermatogenesis with spermatogonia (cells with perinuclear halo near the seminiferous wall), spermatocytes of first order (filamentous nuclei), spermatocytes of second order, and immature spermatids (cells with small nuclei). Mature spermatids (elongated nuclei, some with subnuclear vacuole). |
Spermatogonia (21.4 ± 4 per seminiferous tubule), located near the tubular wall, are placed in rows, with round nuclei, regular chromatin, and a perinuclear halo.
Spermatocytes (31 ± 6 per tubule) of the first order have nuclei with filamentous chromatin because of their involvement in meiosis. The spermatocytes of second order are not recognized because they have a very short average life (about 8 hours),29 and since they are already haploid cells, they have very small nuclei that cannot be distinguished with certainty from the immature spermatids.
Immature spermatids (37 ± 7 per tubule) are cells with central nuclei with lymphoid characteristics and somewhat abundant and clear cytoplasm.
Mature spermatids (25 ± 4 per tubule), the last maturing phase that we can recognize, are located near the tubular lumens and have elongated nuclei with scanty cytoplasm.
The order of these germ cells in the tubule is apparently irregular, but for some time it has been recorded that there are six distinct stages,35 a consequence of a helicoidal ordering throughout the tubule. These stages of spermatogenesis are defined as a characteristic association of germ cells representing several waves of spermatogenesis that occur simultaneously within the seminiferous tubules. Each stage has a specific duration, and the groups always occur in the same order, so that once the complete cycle is finalized it begins again.36 During the maturation process,
from spermatogonium to mature spermatids, all the cells belonging to the same clone are interconnected.37 Stage I is composed of spermatogonia, pachytene primary spermatocytes, and round and elongating spermatids. Stage II includes spermatogonia, pachytene primary spermatocytes, round and elongated spermatids, and residual bodies derived from spermatid cytoplasm within Sertoli cells. Stage III is characterized by the beginning of spermatid nuclear condensation and the entrance of type B spermatogonia into meiosis. Stages IV and V comprise pachytene primary spermatocytes, and can be differentiated by the presence of leptotene and zygotene primary spermatocytes. In Stage V, the secondary spermatocytes undergo a second meiotic division after a very short interphase.
from spermatogonium to mature spermatids, all the cells belonging to the same clone are interconnected.37 Stage I is composed of spermatogonia, pachytene primary spermatocytes, and round and elongating spermatids. Stage II includes spermatogonia, pachytene primary spermatocytes, round and elongated spermatids, and residual bodies derived from spermatid cytoplasm within Sertoli cells. Stage III is characterized by the beginning of spermatid nuclear condensation and the entrance of type B spermatogonia into meiosis. Stages IV and V comprise pachytene primary spermatocytes, and can be differentiated by the presence of leptotene and zygotene primary spermatocytes. In Stage V, the secondary spermatocytes undergo a second meiotic division after a very short interphase.
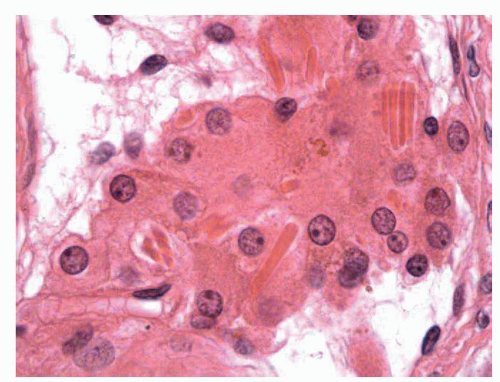 FIGURE 11-4 ▪ Adult testis. Normal Leydig cells around capillary vessels with abundant eosinophilic cytoplasm and Reinke crystalloids. |
In the interstitium, connective tissue collagen fibers with contractile capacity are present. The Leydig cells are the most important component, making up to 5% to 12% of the testicular volume.38 They are polygonal and have PAS-positive, eosinophilic cytoplasm with a central nucleus and prominent nucleolus, grouped in small nests (1 or 2 nest per tubule or 5 ± 0.2 cells per tubule) around the capillaries of the intertubular space. The fetal Leydig cells produce testosterone; immature Leydig cells produce 3α and 17β-diol and adult Leydig cells steroids. Characteristically, some eosinophilic crystalline structures can be found in the cytoplasm (Reinke crystalloids); these are probably subunits of globular proteins whose functional meaning is not known39 (Fig. 11-4). Other cellular elements of the interstitium are the macrophages that secrete interleukin 1, which stimulates the proliferation of the germ cells, and the mast cells.
AGING TESTIS
The changes considered as age-related involution do not have a specific age at commencement, and the fact that we are able to see normal testicles in all age groups indicates that there are marked individual variations.40 The involution seems to be related more to hormonal levels than to age; however, after age 70 years, the changes of atrophy are frequent, and after age 80 years, the testicles of almost all men have a certain degree of fibrosis in the tubular wall, although they can preserve spermatogenesis.40
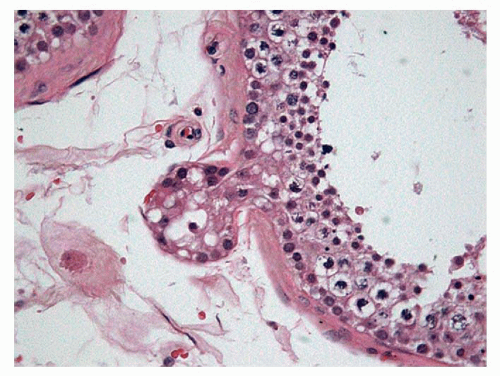 FIGURE 11-5 ▪ Elderly testis. Seminiferous tubule with germ cell atrophy and parietal diverticulum. |
Several authors have observed foci of total tubular sclerosis, suggestive of a local ischemic phenomenon, as well as the appearance of tubular diverticula (Fig. 11-5) directly proportional to age, for possible weakening of the tubular wall together with obstructive phenomenon due to storage of fluid in the Sertoli cells.41 The Sertoli cells store glycogen, ascorbic acid, and lipids, but it is difficult to interpret the significance of this accumulation because it starts at a very early age.42 There can also be a decrease in the number of cells40 in the aged testis and multinucleation in 4% of testicles.43,44
The loss of germ cells begins with the spermatids and progressively affects the predecessor cells,45 with the pale spermatogonia disappearing in about the sixth decade and the dark ones in about the eighth decade.46 A curious finding is the appearance of multinucleated spermatids (from 5 to 86 nuclei) that have been interpreted as an expression of active karyokinesis without cytokinesis, typical of aging.47
In the interstitium, one can observe an increase in the connective tissue earlier than in the tubule, and there are data indicating that this increase is regulated by differential expression of TGF-β proteins and decreased levels of TGF-β2.48 As with the Leydig cells, there are contradictory observations; some authors have reported that their numbers decrease with age, especially in relation to the decrease of LH,40 whereas others have found a certain compensatory increase.49 Other changes that can be attributed to involution are intercellular fibrosis, cytoplasmic microvacuolization with accumulation of lipofuscin, and binucleation.50
ANATOMY OF THE ADULT TESTIS
Gross Anatomy and Microanatomy
The adult testis measures approximately 4 to 5 cm × 3.5 cm × 3 cm and weighs 15 to 19 g with the right usually being 10% heavier that the left. The external surface is covered by a capsule (tunica albuginea), which is a smooth and homogeneous layer measuring 400 to 459 μm in thickness in the adult. The tunica albuginea is composed of three layers. The outermost layer comprises of dense connective tissue and is lined by mesothelium. The middle layer represents less dense fibrous tissue. The inner most layer is rich vascular connective tissue. From this tunica albuginea layer, numerous fibrous septa emerge to divide the testicular parenchyma into approximately 250 lobules, and meet in a solid posterior area at the hilum of the testis, near the epididymis, which is called the mediastinum of the testis.
Each lobule of the testis contains one to four seminiferous tubules. Every tubule has a diameter of 180 μm and a total average length of 540 m. The tubules are a convoluted structure with numerous communications between the arms of the loop. Each arm of the loop empties into the mediastinum.
The mediastinum contains the rete testis, a connecting structure between the seminiferous tubules with the ductuli efferentes in the epididymis. There are around 1,500 units of seminiferous tubules to the rete. It is divided into three parts: the septal portion with the tubulae reti, which are short tubules 0.5 to 1.0 mm in length that connect the two ends of the seminiferous tubules to the mediastinal part with the tunical rete, which is a cavernous network of interconnecting channels between tubular reti and the extratesticular part with the bullae retis, which are vesicular channels measuring 3 mm in width that anastomose together to form the ductuli efferentes. In the mediastinal and external rete testis parts, fibrous columns or strands covered by epithelium called chordae retis are present to connect the different walls.
The rete testis epithelium is composed of flattened cells interspersed with small areas of columnar cells. Both cell types have single centrally located cilia and numerous microvilli on their free surfaces. These cells rest on a basal lamina surrounded by a layer of myofibroblasts and external layer of fibroblasts and collagen and elastic fibers.
Apart from the connection between the testis and the epididymis, the rete testis produces a pressure gradient internally for reabsorption of protein and potassium from tubular fluid.
Blood Supply and Lymphatics
The testis is supplied by the testicular artery, which arises from the abdominal aorta. In the spermatic cord, the testicular artery gives multiple branches that run along the interlobular septa of the testis. These centripetal arteries lead to the mediastinum testis and give off branches called centrifugal arteries.
The inner two-thirds of the testicular parenchyma are drained by veins that follow the interlobular septa to the mediastinum (centripetal veins). The outer third is drained by veins that lead to the tunica albuginea (centrifugal veins). Both centripetal and centrifugal veins join to form the pampiniform plexus, which drains the testis via the spermatic cord.
Lymphatic vessels are poorly developed in the testis and limited to the tunica vasculosa and interlobular septa where they accompany arterioles and venules.
Nerves
Efferent innervation of the testis is mainly supplied by neurons of the pelvic ganglia, where contralateral and bilateral neural connections occur. Postganglionic nerve fibers enter the testis via the pelvic nerves, extend throughout the tunica vasculosa, and follow the interlobular septa to reach the interstitium. The nerve fibers end in the wall of arterioles, the wall of seminiferous tubules, and the Leydig cells. Adrenergic nerve fibers innervate the tunica albuginea and the blood vessels of the tunica vasculosa. Peptidenergic nerve endings are uncommon. Afferent nerve endings from corpuscles similar to those of Meissner and Pacini are observed in the tunica albuginea.
CONGENITAL ANOMALIES
Disorders in Number and Size
Congenital disorders in number and size constitute one of the less frequently seen groups of testicular anomalies.
Monorchidism
Monorchidism is the congenital absence of one testicle. Its incidence is 1 in 5,000 masculine births, with a predominance of the left side (68.7%).29 Among boys with monorchidism, 20% have other congenital genital malformations, and 30% have anomalies of the urinary tract. For a correct diagnosis, the possibility of any evidence of a testicle must be excluded; it is not sufficient to find blind seminal canals. All remnants found at exploration should be removed, and the absence of testicular parenchyma should be confirmed before diagnosing monorchidism. The finding of blindly ending spermatic vessels is the only accepted evidence of monorchidism.16 The contralateral testicle can present a compensatory hypertrophy that can double the volume of the normal testicle; this is a consequence of the change in endocrine feedback that increases the FSH.51 The increase in testis weight is correlated, in experimental models, with an increase in total seminiferous tubule length and a larger cross-sectional area, which is due in part to the greater number of germ cells per testis.52
Anorchidism
Although the term anorchidism strictly refers to the total absence of both testicles, a series of pathologic situations is usually included in this term that varies from the actual lack
of gonads to extreme hypoplasia or very prolonged concealment of the testicles, for which reason the term testicular regression syndrome53 has been created and includes the following disorders:
of gonads to extreme hypoplasia or very prolonged concealment of the testicles, for which reason the term testicular regression syndrome53 has been created and includes the following disorders:
True Agonadism
True agonadism is a genuine absence of testicles in 46XY patients with ambiguous external genitals, and more rarely, in 46XX patients with external feminine genitals, with the possibility of there being a rudimentary uterine tube. The cause is unknown, although there are cases associated with heterozygote mutation of gene WT1.54 This disorder can be associated with various syndromes of multiorganic malformations.55
Rudimentary Testes Syndrome
Rudimentary testes syndrome is the presence of cryptorchidic rests of testicles with occasional seminiferous tubules in which Sertoli cells and some spermatogonia are present. The external genitals are not ambiguous but are hypoplastic (micropenis).56
Congenital Bilateral Anorchidism
Congenital bilateral anorchidism is the strict bilateral absence of testicles with only wolffian elements and without müllerian rests. The phenotype is normal masculine or hypoplasia, and its incidence is 1 in 20,000 male births. The cause is unknown; there have been attempts to find mutations of the SRY gene, but convincing proof has not been found.57
Vanishing Testes Syndrome
Vanishing testes syndrome is the disappearance of the testicles from the last months of pregnancy until puberty; this disorder should be considered only as a prolonged concealment of the testicles, since they can be found in the inguinal canal or in the upper scrotum. The deep alterations of these testicles, represented only by occasional seminiferous tubules accompanied by epididymis, are more attributable to perinatal scrotal torsion than to genetic causes.55
Leydig-cell-only Syndrome
Leydig-cell-only syndrome is characterized by finding only clusters of functioning Leydig cells in the spermatic cords, with sufficient testosterone for male phenotype but insufficient for the complete development of secondary sex characteristics.
Sinorchidism
Sinorchidism is an extremely infrequent anomaly, characterized by the fusion of both testicles, each with their respective epididymis located in the midline. This fusion is usually associated with other fusions such as those of the adrenal glands and horseshoe kidney.29
Polyorchidism
Polyorchidism is the presence of more than two testicles, with three being the most frequent. This anomaly occurs infrequently, and the embryologic origin is not clear; the longitudinal division of all the structures of the genital ridges and mesonephric ducts with only the longitudinal division of the genital ridges, and the high transverse division of the genital ridges and the low transverse division of the genital ridges have been proposed. The extra testicle is frequently intrascrotal and many times presents diverse alterations of spermatogenesis. There is no reason for a greater incidence of malignant transformation, although some cases have been reported with germ cell tumors.58,59
Macroorchidism
An increase in volume of testicular parenchyma can correspond to diverse pathologic situations. Some of them are consequences of other pathologic conditions, such as the loss of total testicular parenchyma (e.g., the aforementioned compensatory hypertrophy) or the secretion of androgens by Leydig cell tumors; but others are considered true intrinsic anomalies, such as the following:
Idiopathic Benign Macroorchidism
Idiopathic benign macroorchidism is characterized by an increase in the longitude of the tubules. It is caused perhaps by the greater sensitivity of hormonal receptors, which in the development of these testicular alterations are curiously found during spermatogenesis.60
Precocious Puberty
For practical purposes, this is considered to be before 8 years of age in girls and 9 years in boys. The incidence is estimated at between 1 in 5,000 and 1 in 10,000, with a female:male ratio higher than 20:1. In boys, the first symptom is rapid testicular enlargement followed by growth of pubic and axillary hair, enlargement of the penis, and acceleration of skeletal growth.
Precocious puberty results from the early differentiation of Leydig cells, with complete spermatogenesis and abnormal spermatids and, in the absence of stimulus, by pituitary gonadotropin (familial testotoxicosis)61,62; it can also result from alterations of the central nervous system such as those that occur in McCune-Albright or von Recklinghausen syndromes.29 Other causes are the Leydig cell tumors and congenital adrenal hyperplasia.
Other Macroorchidisms
Other alterations such as fragile X chromosome (Martin-Bell syndrome)63 or congenital Leydig cell hyperplasia can also be accompanied by macroorchidism because of the transfer of human chorionic gonadotropin from the mother to the fetus, similar to what occurs in diabetic mothers with hypertension.64,65 In bilateral megalotestes with low gonadotropin, with excellent fertility parameters despite the unusually low hormone levels, no specific pathology underlying the large gonadal volume could be identified.66
Alterations of Location
Some congenital anomalies can be classified by their locations outside the normal path of testicular descent (ectopia) or in the path of descent (undescended testes). The location
in the superficial inguinal pouch is considered to be ectopia by some researchers or to be cryptorchidism by others.
in the superficial inguinal pouch is considered to be ectopia by some researchers or to be cryptorchidism by others.
Ectopia
There are two types of ectopia: one that involves the complete testicle and the other that involves parts of the testicular parenchyma.
Complete Ectopia of the Testicle
Complete ectopia of the testicle occurs when the testicles complete the transinguinal migration and then divert to another location under the superficial inguinal ring. This anomaly is considered to be related to alterations in the gubernaculum testis and its branches since the ectopias that can be observed (suprapubic, superficial inguinal, femoral, transverse scrotal, and perineal)67 correspond to the sites of these branches. These testicles have normal characteristics and are not associated with a greater incidence of neoplasia.52
Testicular Parenchymal Ectopia
Seminiferous tubule ectopia is characterized by the presence of seminiferous tubules in a normal albuginea, in contiguity with the tubules of the parenchyma, without evidence of ovarian stroma (fundamental to distinguish it from some testicular dysgenesis), and with clear delimitation between the tunica albuginea and the testicular parenchyma.68
Leydig cells ectopia can be located in the testicle (interlobular septa, rete testis, tunica albuginea, or complete fibrotic tubules) or in extratesticular structures such as the epididymis or spermatic cord (generally perineural). These cells seem to be less functional than do those with a normal location. Their origin is not clear, and cell migrations can be assumed since cells do not display ectopic differentiation.69, 70, 71
Undescended testis (Cryptorchidism)
Undescended testes are the most common testicular anomaly. Scrotal testicular absence is seen in 30.3% of premature boys, and incomplete descent is seen in 3.2% of full-term boys. In most boys with incomplete descent, the testes will descend spontaneously within the first 3 months; by the end of the 1st year, the testes will not have descended in only 0.8%. Spontaneous resolution is rare after the 1st year.72
True Cryptorchidism
In boys with cryptorchidism, the testicle remains immovable along some areas of the testicular descent path; this occurs in 25% of cases of empty scrotum. It can be associated with complex syndromes (e.g., Klinefelter, Kallmann, or Noonan) or with other malformations such as omphalocele and myelomeningocele.
The mechanism for testicular descent involves participation by hormonal and mechanical factors and although it is not completely clear, the process involves three stages viz. nephric displacement (7th week), transabdominal descent (12th week), and inguinal descent (between 7th month and birth).73
The gubernaculum testis regulates testicular descent along with the formation of the inguinal canal and processus vaginalis.74 It is a complex process involving a normally functioning hypothalamo-pituitary-gonadal axis, normal development of abdominal wall musculature, gubernaculum, and the processus vaginalis,75 along with a normally functioning endocrine system of the testis.
The hormonal requisites for testicular descent are varied and not entirely elucidated. One of the most essential factors for testicular descent is insulin-like factor-3 (INSF-3), which is produced by Leydig cells and is independent of the androgen pathway. It stimulates gubernacular swelling, which is required for the initiation of testicular descent.76 INSF-3 gene mutations or mutations of its receptors GREAT (G-protein-coupled receptor affecting testicular descent) or LGRB-8 (leucine-rich repeat-containing G-protein-coupled receptor-8) result in improper testicular descent and cryptorchidism.77 Gubernaculum swelling also involves contributions from AMH and androgens.
Inguinoscrotal descent has not been fully explained; however, androgens and the genitofemoral nerve are two significant factors behind the process. Androgens act on the nucleus of the genitofemoral nerve in the spinal cord as opposed to direct action on the gubernaculums testis, effectively bringing about the masculinization of the neurons comprising the nucleus78 accompanied by secretion of large amounts of calcitonin gene-related peptide (CGRP). In turn, CGRP acts on the cremasteric muscle that develops in the gubernaculum and is innervated by the genitofemoral nerve. This theory is supported by the fact that neurogenic atrophy of this muscle is seen in cryptorchid patients.79
A host of other factors are involved in testicular descent including epidermal growth factor (EGF) and estrogens. Experimental studies have demonstrated that estradiol decreases gubernacular swelling and plays a role in stabilizing müllerian ducts. One of the proposed hypotheses states that cell proliferation resulting in gubernacular swelling is inhibited by estradiol.80 EGF is involved in testicular descent by its action at various sites along the entire gonadal-placental axis. EGF levels in maternal circulation rise immediately prior to fetal masculinization.81 The placenta has an increased concentration of EGF receptors, and placental stimulation by EGF may cause hCG production. This chain of events may also stimulate androgen production by Leydig cells and a summation of these might bring about testicular descent.
The gubernaculum and processus vaginalis regress upon birth and the gubernaculum is replaced by fibrous tissue comprising the scrotal ligament. The processus vaginalis atrophies along its cephalic portion after testicular descent. If this process is exaggerated, a testis that descended normally may be caused to ascend and lead to a cryptorchid condition.82
The pathogenesis of testicular alteration is very controversial, and opinions vary from those who believe the alterations are based on immunologic changes (due to the
alteration of the hemato-testicular barrier because of hyperthermia)83 to those who believe that the alterations are a result of dysgenetic expression. Independent of the etiopathogenetic hypotheses, the important consideration is to know the morphologic variations, their chronology, and the possibility of preventing them.
alteration of the hemato-testicular barrier because of hyperthermia)83 to those who believe that the alterations are a result of dysgenetic expression. Independent of the etiopathogenetic hypotheses, the important consideration is to know the morphologic variations, their chronology, and the possibility of preventing them.
We can systematize the testicular changes of cryptorchidism by age groups, as follows:
Changes in Prepubertal Cryptorchid Testes.
In patients with prepubertal cryptorchid testes, 74% of the testes showed severe decrease in the average tubular diameter, marked decrease of the tubular fertility index, and hyperplasia of the Sertoli cells.84 These findings, together with the frequent alteration of the contralateral testicle, reinforce the hypothesis that some cases represent authentic testicular dysgenesis. Other changes such as annular tubules and calcospherites (for probable cellular peeling and subsequent calcification)85 are more difficult to explain.
Changes in Pubertal Cryptorchid Testes.
During puberty in patients with pubertal cryptorchid testes, the lesions are very deep, and almost all patients experience some of the anomalies already described in the prepubertal cryptorchidic testes.86 This disorder appears to be associated with progressive worsening of the structures until becoming terminal, typically at 13 years of age.87
Changes in the Adult Cryptorchid Testes.
In adult men with cryptorchid testes, the entire testicle shows advanced atrophic changes88 (Fig. 11-6). In a series by one of the authors (F.A.), changes in the cryptorchid testes that descended at prepubertal ages showed structural normality in only 7.7% of patients, and fibrotic changes in 46% of the cases.
Several foci of infantile (immature) seminiferous tubules can be present. Each group of tubules appears well delimited but unencapsulated. Nodule size varies from microscopic to 5 mm. On cut section, each nodule is distinguished by its whitish color. The seminiferous tubules have a prepubertal diameter and may be anastomotic. The epithelium is columnar or pseudostratified, devoid of lumina, and usually consists only of Sertoli cells. The cells have elongated hyperchromatic nuclei with one or several peripherally placed small nucleoli. The interstitium varies from scant to well collagenized. Leydig cells are usually absent in these areas and, if present, their numbers are low. Sertoli cell nodule is found in most adult cryptorchid testes, regardless of when the testes descended. It is also present in 22% of normal scrotal testes in some series and is an occasional finding in males with idiopathic infertility.
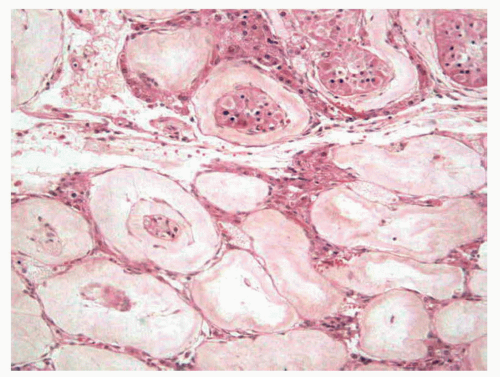 FIGURE 11-6 ▪ Adult cryptorchid testis. Advanced tubular wall fibrosis with complete absence of tubular cells or only residual Sertoli cells. |
Much has been discussed on the use of orchiopexy in improving fertility, but its success has not been proven.86 Also not proven is the justification for a systematic biopsy of the cryptorchid testis to predict its functional capacity or to detect an intratubular germ cell neoplasia. One of the most debated subjects is the incidence of a germ cell tumor in a cryptorchid testis since the risk of developing germ cell tumors is 4 to 10 times higher than in a normally descended testicle.89 Seminoma is the most common histologic type. Orchiopexy does not decrease the incidence of neoplasia, which supports the hypothesis that changes of these testicles are dysgenetic.
Obstructed Testes
The testes are located superficially in the inguinal Denis-Browne pouch. Some authors consider them as ectopic and others as true cryptorchidism. The morphologic changes are similar to those in cryptorchidism.
Retractile Testes
The testicle may ascend to the scrotum at the time of exploration. Some alterations such as variable germ cell atrophy can be present from one lobule to the other.90
Gonadal/Testicular Dysgenesis
Gonadal dysgenesis is characterized by a feminine phenotype with amenorrhea and hypoplasia of the uterus and fallopian tubes. Dysgenesis is usually classified according to karyotype and therefore can be
46XY GONADAL DYSGENESIS (Swyer syndrome) is characterized by a female phenotype without signs of the Turner syndrome with infantilism. It is possible to find fused labia majora, hypertrophic clitoris, and hypospadias. Some patients have mental retardation and chronic renal insufficiency. The typical gonads are the fibrous streak (see below).
46XX GONADAL DYSGENESIS with normal genitals and ovarian hypoplasia (see below) rather than streak gonads.
45XO GONADAL DYSGENESIS with stigmata of the Turner syndrome; the external genitalia are female and infantile. The typical streak gonads are present.
MIXED DYSGENESIS with streak gonads in some cases associated with testis.
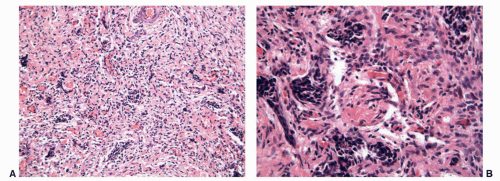 FIGURE 11-7 ▪ Gonadal dysgenesis. Fibrous streak with ovarian-like stroma (A) and occasional rete testis like channels (B). |
The streak gonads may consist only of ovarian stroma with a nodular pattern (typical of 46XY, and 45XO), fibrous tissue with occasional ovarian follicles (especially in 46XX/45XO), or fibrous tissue with tubules and channels resembling rete testis (in 46XY)55,91 (Fig. 11-7).
The hypoplasic ovaries, in 46XX gonadal dysgenesis are characterized by small ovaries that are more often hypoplastic and rarely streak gonads. The histologic picture of the ovaries consists of fibrous stroma without generative elements or with a small number of primary follicles, but sometimes also with a single growing graafian follicle.92
The incidence of tumors in gonadal dysgenesis is variable. Approximately 25% to 30% of patients with 46XY dysgenesis can develop gonadoblastomas, seminomas, or other germ cell tumors (Fig. 11-8), for which preventive extirpation is recommended.93 In mixed dysgenesis, tumors develop in 25% of the cases94; in the other forms, the incidence of tumors is lower.55
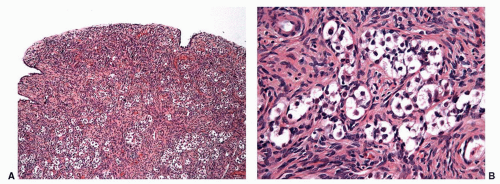 FIGURE 11-8 ▪ Gonadoblastoma in gonadal dysgenesis. (A) Low power view; (B) high magnification. Large germ cells with clear cytoplasm (seminoma) surrounded by small cells resembling immature Sertoli cells and granulosa cells. |
In mixed dysgenesis, along with a fibrous streak, testicular dysgenesis can be found that is characterized by a central testicular area comprising of seminiferous tubules that are smaller than normal but identifiable, surrounded by ovarian stroma and branched tubules with an albuginea that may differ by the absence of tunica vasculosa.55
True Hermaphroditism
The term hermaphroditism should be applied only to patients who have both testicular and ovarian tissue (Fig. 11-9). It is a pathologic entity with a difficult clinical diagnosis. In patients with a masculine phenotype, hermaphroditism can often be recognized only in puberty by developing gynecomastia, which is present in nearly all of these patients29; in those who have a feminine phenotype, clitoromegaly or irregular menstruation can indicate this condition.
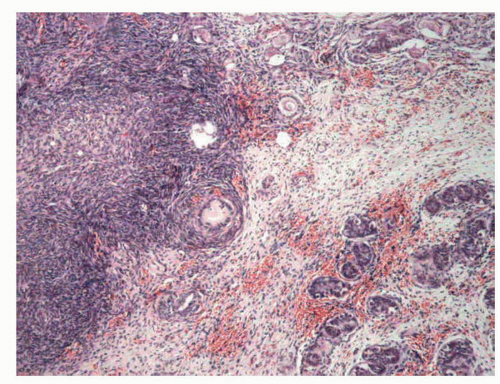 FIGURE 11-9 ▪ True hermaphroditism. Ovotestis with ovarian tissue in upper left area and seminiferous tubules in lower right area, separated by nonspecific stromal tissue. |
The gonads can be any type of combination of both tissues, but in 44.4% of cases it constitutes ovotestis. In about half of the cases, the location is intra-abdominal. In the rest of the cases, the location is inguinal, scrotal, or labial. Only 5% of patients with ovotestis are bilateral and the remainder are unilateral, with a predominance of the right side.95 The ovotestis can be (a) bilobated, with one of the tissues having a pedicle and in each of the lobes, (b) ovoid, with the central testicular parenchyma and the ovarian tissue around it,29 or (c) intermixed (occasionally), with both ovocytes and seminiferous tubules.55 After puberty, seminiferous tubules remain small and often contain dysgenetic Sertoli cells similar to cryptorchid testis. Incomplete spermatogenesis has been reported, but complete spermatogenesis is very rare. The ovary is most frequently on the left side and usually hypoplastic with few primordial follicles. Occasionally, it is functionally and histologically normal.
About 4.6% of ovotestes develop germ cell tumors96,97 and the most frequent lesions are gonadoblastoma and dysgerminoma/seminoma, followed by yolk sac tumors, mature teratoma, and carcinoid tumors. These tumors can grow to a large size. The testicle must be removed, and the residual gonad should be monitored by regular sonography explorations, particularly in patients with chromosomal mosaicisms.
Male Pseudohermaphroditism
Any process that alters any of the mechanisms for the correct expression of masculine differentiation can result in a state of male pseudohermaphroditism (Table 11-2). Situations inducing male pseudohermaphroditism include the following:
Alterations in Leydig Cell Activity
Alterations in Leydig cell activity are associated with deficiencies in androgen synthesis or with deficient formation of pregnenolone, 3β-hydroxysteroid dehydrogenase, 17α-hydroxylase, 17.20 desmolase, or 17β-hydroxysteroid dehydrogenase.29
Table 11-2 ▪ CAUSES OF MALE PSEUDOHERMAPHRODITISM | |||||||
|---|---|---|---|---|---|---|---|
|
Insufficient secretion of testosterone also can be motivated by Leydig cell hypoplasia




Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
