Our better understanding of the morphologic spectrum of renal cortical tumors has resulted in a clinically more relevant classification of these tumor types. We now recognize that “granular cell” and “sarcomatoid” renal cell carcinoma are only nonspecific descriptors, and that such features are seen in a variety of types of renal tumors. The authors believe that the recently gained knowledge about molecular-driven antigen expression will play an important role in the characterization, development, and evaluation of targeted therapies in kidney cancer in the coming years.
Until the 1980s, renal cortical tumors had been traditionally classified as clear cell, granular cell, papillary and sarcomatoid carcinoma, or renal oncocytoma. In 1985, Thöenes and colleagues first described chromophobe renal cell carcinoma (RCC) in human beings, followed soon by its eosinophilic variant. This led to a reassessment of the morphologic classification of renal tumors and laid the framework for the clinicopathologically more valid classification used today ( Box 1 ). We know now that “granular cell carcinoma” and “sarcomatoid carcinoma” are not specific entities, and that granular and sarcomatoid features can be seen in a variety of renal tumors with markedly diverse biologic potential. The current histologic classification has also been validated by a number of molecular studies. The implications of this now universally accepted classification system are that publications about clinicopathologic aspects of renal tumors from the pre-1985 era may not correspond to the current knowledge about these tumors.
Benign
Renal oncocytoma
Papillary adenoma
Metanephric adenoma
Metanephric adenofibroma
Malignant
Clear Cell (conventional) RCC
Papillary RCC
Chromophobe RCC
Collecting duct carcinoma
Medullary carcinoma
Tubulocystic RCC
Acquired cystic disease of kidney-associated RCC
RCC, unclassified
Mucinous tubular and spindle cell carcinoma
Translocation associated carcinomas
Tumors of indefinite malignant potential
Multilocular cystic RCC
The better understanding of the molecular aspects of renal tumors in the recent past has resulted in the realization that differing molecular pathways are involved in different renal tumors. This has resulted in the development and usage of multiple targeted therapies, particularly for advanced clear cell RCC, with promising initial results. At the same time, this knowledge has provided pathologists with additional tools to investigate molecular pathway markers in different subtypes of kidney tumors, and to potentially use them for their differential diagnostic and prognostic evaluation. Therefore, the challenges to the genitourinary pathologists are to ( i ) understand the morphologic diversity seen in different tumors to classify them more precisely, ( ii ) understand the antigenic diversity in these tumors and its application to targeted therapy, and ( iii ) be able to classify these tumors on minimal material, such as needle biopsies and aspirates.
Grading and staging of renal tumors
Among the several grading schemes proposed for renal tumors over the years, Fuhrman’s system is the most widely used. It uses nuclear grades based on nuclear size, irregularity of the nuclear membrane, and nucleolar prominence. For practical purposes, easily identifiable nucleoli at low magnification (10×) examination are characteristic of nuclear grade 3 or 4, with grade 4 nuclei also showing marked pleomorphism and multilobulation. Grade 1 or 2 nuclei generally require examination at high magnification (40×) to identify and evaluate the level of prominence of the nucleoli. While the utility of Fuhrman’s grading system in clear cell RCC is well established, its value in papillary and chromophobe RCC remains controversial. Renal oncocytomas are not graded, as they are benign tumors.
The TNM classification of the American Joint Committee on Cancer (AJCC) and Union Internationale Contre le Cancer is the most widely used and clinically relevant staging system for renal tumors. Over the years it has been modified and improved multiple times; the latest modifications were made in 2002. While it cannot claim to be a perfect staging system, at present the usage of TNM classification is recommended in the hope of establishing uniformity in reporting, while future AJCC staging criteria will almost certainly correct whatever deficiencies are realized in the present system.
Recently, a series of publications have highlighted the importance of the renal sinus and sinus veins in the staging of RCC, factors first included in the AJCC staging system in 2002. In a retrospective study, it was recently shown that a significant proportion of cases that die of apparent pT1 disease in fact have renal sinus fat invasion. Therefore, a careful gross examination and adequate sampling of the renal sinus-tumor interface, especially in larger tumors, is strongly recommended.
Clear cell (conventional) RCC
Clear cell RCC comprises approximately 60% to 65% of all renal cortical tumors. They typically show tumor cells with clear cytoplasm (because of the abundant intracytoplasmic lipid and glycogen) and an acinar or solid-nested growth pattern, invested by an intricate, arborizing vasculature. The cytoplasmic clarity is especially more common in low-grade tumors. The higher grade tumors less often show pure clear cell cytology, and may contain variable proportions of (or even exclusive) tumor cells with granular/eosinophilic cytoplasm ( Fig. 1 A ). In these high-grade areas, a loss of the acinar growth pattern is quite frequent, and such areas are more likely to have solid or sometimes sarcomatoid histology. It is important to look for areas of transition to a lower grade so as to establish the correct diagnosis. The intricate vasculature tends to be retained in most tumors, except in the very high-grade areas with solid or sarcomatoid differentiation. Sarcomatoid differentiation occurs in approximately 5% of these tumors and, as in other subtypes of RCC, is an indicator of high-grade tumor with generally ominous prognosis. These tumors may take on a papillary or pseudopapillary appearance focally, but this is usually because of degenerative changes rather than true papillae formation.
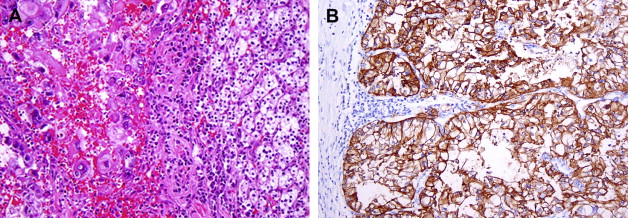
Clear cell RCCs are characterized by the loss of genetic material of the short arm of chromosome 3 (3p) and mutations in the von Hippel-Lindau tumor suppressor ( VHL ) gene. In patients with von Hippel-Lindau disease, such losses and mutations are described in virtually all cases. Somatic mutations/promoter hypermethylations in the same region can be found in 75% to 80% of the more common sporadic tumors as well. The product of a normal VHL gene, pVHL, is required to target and degrade hypoxia-induced factor (HIF) in normoxemic states. In cells that are hypoxic, or lack pVHL (as in most cases of clear cell RCC), HIF escapes degradation and activates downstream targets, including vascular endothelial growth factor, platelet derived growth factor, glucose transporter 1, and carbonic anhydrase IX (CA IX), among others. The expression of these downstream products at the immunohistochemical level may be used as diagnostic markers of clear cell RCC ( Fig. 1 B) or as targets for novel therapies. CA IX and HIF-1 immunohistochemical expression has also been reported to be related to prognosis, although large prospective studies evaluating such associations are currently not available.
Clear cell (conventional) RCC
Clear cell RCC comprises approximately 60% to 65% of all renal cortical tumors. They typically show tumor cells with clear cytoplasm (because of the abundant intracytoplasmic lipid and glycogen) and an acinar or solid-nested growth pattern, invested by an intricate, arborizing vasculature. The cytoplasmic clarity is especially more common in low-grade tumors. The higher grade tumors less often show pure clear cell cytology, and may contain variable proportions of (or even exclusive) tumor cells with granular/eosinophilic cytoplasm ( Fig. 1 A ). In these high-grade areas, a loss of the acinar growth pattern is quite frequent, and such areas are more likely to have solid or sometimes sarcomatoid histology. It is important to look for areas of transition to a lower grade so as to establish the correct diagnosis. The intricate vasculature tends to be retained in most tumors, except in the very high-grade areas with solid or sarcomatoid differentiation. Sarcomatoid differentiation occurs in approximately 5% of these tumors and, as in other subtypes of RCC, is an indicator of high-grade tumor with generally ominous prognosis. These tumors may take on a papillary or pseudopapillary appearance focally, but this is usually because of degenerative changes rather than true papillae formation.
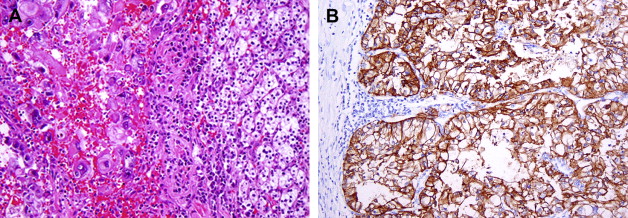
Clear cell RCCs are characterized by the loss of genetic material of the short arm of chromosome 3 (3p) and mutations in the von Hippel-Lindau tumor suppressor ( VHL ) gene. In patients with von Hippel-Lindau disease, such losses and mutations are described in virtually all cases. Somatic mutations/promoter hypermethylations in the same region can be found in 75% to 80% of the more common sporadic tumors as well. The product of a normal VHL gene, pVHL, is required to target and degrade hypoxia-induced factor (HIF) in normoxemic states. In cells that are hypoxic, or lack pVHL (as in most cases of clear cell RCC), HIF escapes degradation and activates downstream targets, including vascular endothelial growth factor, platelet derived growth factor, glucose transporter 1, and carbonic anhydrase IX (CA IX), among others. The expression of these downstream products at the immunohistochemical level may be used as diagnostic markers of clear cell RCC ( Fig. 1 B) or as targets for novel therapies. CA IX and HIF-1 immunohistochemical expression has also been reported to be related to prognosis, although large prospective studies evaluating such associations are currently not available.
Papillary RCC
Papillary RCC (PRCC) constitute up to 15% of all renal cortical neoplasms. There are at least two distinct types of papillary RCC, both at the morphologic and genetic level, and they appear to have distinct clinical behaviors as well. Architecturally, most of these tumors have a papillary, tubular, or tubulo-papillary growth pattern. Some have asolid growth pattern because of compression of the papillary structures, while others show a glomeruloid appearance. The cytoplasm may be amphophilic, eosinophilic, or even partially clear. Papillary RCCs are often multifocal, are frequently associated with papillary adenomas (tumors less than 5 mm in size, by definition) and are the most common RCC type with bilateral disease. Classically, papillary tumors display abundant lipid laden, foamy macrophages within fibrovascular cores, a feature helpful in establishing the correct diagnosis. It needs to be emphasized that correct classification of these tumors requires experience and a detailed review of all available slides.
Some experts feel that the Fuhrman grading scheme is well suited for papillary RCC but others disagree. Delahunt and Eble have suggested a two-tiered system (types 1 and 2) ( Fig. 2 ) based on nuclear features and growth pattern characteristics, which has also been accepted in the latest World Health Organization classification. Whether these tumors should be graded and, if so, what system to use remains controversial.
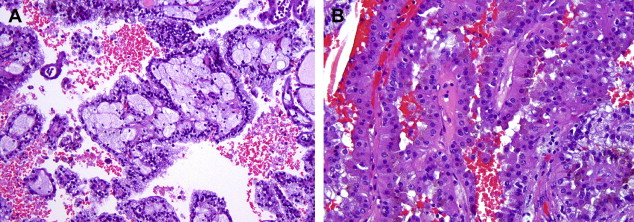
Cytogenetic and molecular studies have revealed distinct findings in PRCC that distinguishes them from other renal epithelial tumors. The majority of tumors, particularly type 1, are characterized by trisomy of chromosomes 7 and 17, as well as loss of chromosome Y. Approximately 10% of the sporadic PRCC also show somatic mutations in the c-MET gene, a genetic abnormality that is common as a germline mutation in familial cases. Some investigators have suggested that tumors exhibiting trisomy 7/17 only are likely to be benign, whereas those exhibiting additional genetic abnormalities will behave aggressively, a hypothesis that has not been confirmed in the literature. Recently, a group from the National Institutes of Health described mutations in the fumarate hydratase gene associated with a subset of type 2 papillary RCC. In most large well-studied series, papillary RCC is a less aggressive tumor than clear cell RCC, with 5-year survival rates of 80% to 85%.
Translocation associated RCC
Translocation associated RCC constitute a small group of RCCs that may have a prominent papillary architecture, often associated with clear cell cytology and psammomatous calcifications. However, nested architecture and cells with granular eosinophilic cytoplasm are also common. In general, they tend to present in younger patients, although a series of cases in adults has also recently been described. These are often associated with an aggressive behavior, more so in the adults. Most cases are associated with t(X;17) or t(X;1), translocations resulting in the fusion of the TFE3 gene on Xp11.2 to usually the ASPL gene on chromosome17 or PRCC on chromosome 1. Both types show strong nuclear immunohistochemical staining with TFE3 antibody. Another subtype seen in children and young adults is characterized by t(6;11) (p21;q12), and it shows strong immunopositivity for transcripton factor TFEB. It is very likely that translocation associated carcinomas were incorrectly classified in the past as unusual variants of clear cell carcinoma, PRCC, or even RCC, unclassified type.
Chromophobe RCC
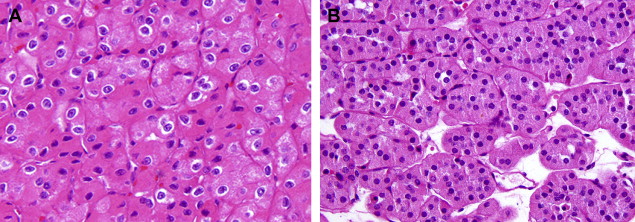
Chromophobe RCC, first described in 1985 by Thöenes and colleagues, constitute approximately 6% of renal cortical neoplasms. Because of their cytoplasmic features, many of these tumors may previously have been classified as clear cell or granular RCC. The morphologic features include a solid growth pattern with sheets of cells separated by incomplete fibrovascular septations. Occasionally, tubular, nested or cord-like growth, mimicking renal oncocytoma, may be present. The cells may be predominantly clear (with finely reticulated cytoplasm), eosinophilic, or most often with a mixture of clear and eosinophilic cells. Nuclei usually exhibit widespread hyperchromasia and nuclear membrane irregularity, although such nuclear features may sometimes be only focal. Classically, one sees perinuclear clearing (perinuclear “halo”), corresponding to distended microvesicles—a distinctive ultrastructural feature of chromophobe RCC. Thöenes and colleagues also described an eosinophilic variant that may be confused with oncocytoma ( Fig. 3 A ). As in other RCC types, chromophobe RCC with sarcomatoid features is a highly aggressive variant.
Chromophobe RCC are genetically characterized by multiple chromosomal losses, usually affecting chromosomes 1, Y, 6, 10, 13, 17, and 21, with resultant hypodiploidy. These tumors have a much better prognosis than clear cell and papillary RCC, with the 5-year disease free survivals of greater than 90%. Even in metastatic settings, Motzer and colleagues demonstrated that chromophobe carcinomas progress more indolently than other nonclear cell RCCs.
Renal oncocytoma
Oncocytomas of the kidney constitute approximately 6% to 9% of renal cortical neoplasms. They are benign neoplasms that require distinction from a number of malignant renal epithelial neoplasms with eosinophilic cytoplasm. Distinction from eosinophilic variant of chromophobe RCC may be particularly difficult for pathologists with less experience. Histologically, oncocytomas are characterized by cells with deeply eosinophilic cytoplasm arranged in nests, cords, or tubules and with uniform round, vesicular nuclei, often with prominent central nucleoli ( Fig. 3 B). Electron microscopy reveals cytoplasm loaded with mitochondria, which is responsible for the cytoplasmic eosinophilia and a mahogany brown gross appearance. Uniformity of nuclear features is the rule, although occasional isolated or groups of cells may exhibit marked degenerative-appearing hyperchromasia and pleomorphism. Prominent papillary growth and extensive tumor necrosis are not the features of renal oncocytoma. Likewise, mitotic activity is rarely noted. A central, stellate scar has been considered characteristic; however, such scars can also be seen in other low-grade renal cortical neoplasms. Microscopically, the scar may contain occasional entrapped tumor cells exhibiting focal cytoplasmic clearing. Other than in this scenario, clear cells are not a feature of renal oncocytoma. Tumor cells may infiltrate perirenal soft tissue, and may occasionally be present within small and even the larger vessels. None of these features affect its benign clinical behavior. Although rare questionable reported cases have metastasized, no known case has died of the disease.
Genetically, oncocytomas do not exhibit 3p-, trisomy 7/17, or multiple combined chromosomal losses. They commonly exhibit loss of chromosomes Y and 1, and a few cases have been described with translocations involving chromosome 11.
Recently, the authors have described a group of patients with multiple oncocytic lesions (oncocytosis). Among several characteristic morphologic features, some had a hybrid morphology between oncocytomas and chromophobe RCC, suggesting that these tumors may be genetically or causally related. In fact, there is a hypothesis that chromophobe tumors may represent a genetic/morphologic progression from oncocytoma. It is very likely that many, if not all of those patients, belonged to Birt-Hogg-Dubé (BHD) syndrome families.
Collecting duct and medullary carcinoma
Collecting duct (Bellini duct) carcinoma (CDC) is very rare, constituting approximately 1% of renal epithelial tumors. In general, the tumors are centered in the renal medulla, particularly when small, and show marked invasive multinodular growth pattern with desmoplasia, highly atypical cytologic features, and papillary, tubular, solid, or microcystic architecture. The papillae rarely, if ever, contain foamy macrophages, but acute or chronic inflammatory cells may be abundant within the tumor ( Fig. 4 ). Characteristically, dysplastic or neoplastic cells may be present within adjacent renal tubules. Cytoplasmic and luminal mucin is frequent. Most show immunoreactivity for carcinoembryogenic antigen, peanut lectin agglutinin, and ulex europaeus agglutinin. Cytokeratins 34BE12 and CK 7 may be positive as well.
Related posts:
 Molecular Biology of Renal Cortical Tumors
Molecular Biology of Renal Cortical Tumors
 Epidemiology, Clinical Staging, and Presentation of Renal Cell Carcinoma
Epidemiology, Clinical Staging, and Presentation of Renal Cell Carcinoma
 Molecular Imaging of Renal Cell Carcinoma
Molecular Imaging of Renal Cell Carcinoma
 Renal Tumor Natural History: the Rationale and Role for Active Surveillance
Renal Tumor Natural History: the Rationale and Role for Active Surveillance
 Choice of Operation for Clinically Localized Renal Tumor
Choice of Operation for Clinically Localized Renal Tumor
 Lymph Node Dissection in the Management of Renal Cell Carcinoma
Lymph Node Dissection in the Management of Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


