The last 10 years have witnessed a dramatic evolution in our understanding of renal cell carcinoma (RCC) biology, which has led to the development of novel medical therapies and revolutionized the approach to their clinical management. This review considers the genetic basis of RCC and the molecular mechanisms of the hypoxia-induced pathway, the mammalian target of rapamycin pathway, the extracellular signal-regulated kinase pathway, and the ubiquitin-proteasome pathway. All these molecular pathways are involved in RCC biology, tumorigenesis, and progression, and serve as the source of new rational treatment strategies based on the design of small molecule inhibitors directed against their targets.
Renal cell carcinoma (RCC) accounts for approximately 3% of all adult malignancies. The incidence of RCC has been steadily increasing; in 2008, over 50,000 Americans will be newly diagnosed with RCC and over 12,000 will die of the disease. The clinical management of RCC is rapidly evolving along with our understanding of the disease process. Historically, RCC was regarded as a single entity. Today, RCC is more accurately recognized as a family of cancers that results from distinct genetic abnormalities that have unique morphologic features but is uniformly derived from renal tubular epithelium. The current World Health Organization classification distinguishes clear cell, papillary, chromophobe, collecting duct, medullary, and unclassified RCC and other rare entities. Advances in genetics and molecular biology have provided insight into the genetic alterations underlying the various renal cortical tumor types and the subsequent downstream molecular pathways involved in their tumorigenesis. These recognized differences reflect a greater sophistication in tumor analysis based on cytology, histology, genetic aberrations, glycogen content, electron microscopy of cytoplasmic microvesicles, and immunohistochemistry of intermediate filament proteins. For example, the discovery of the von Hippel-Lindau ( VHL ) tumor suppressor gene and the hypoxia-induced pathway in clear cell RCC has provided a valuable substrate for application of new strategies for diagnosis, patient selection, and targeted therapy. This article briefly reviews the major genetic alterations underlying our current understanding of renal cortical tumors, addresses the crucial biologic pathways that become altered in the various RCC subtypes ( Fig. 1 ), and gives some consideration to how an understanding of these key molecular receptors and ligands has permitted the rational and evidence-based development of pharmaceutic agents capable of specifically targeting key steps in these pathways.
Hereditary kidney cancer
Hereditary kidney cancer accounts for approximately 3% to 5% of RCC cases. Four well-described forms are distinguished, including VHL, hereditary papillary RCC (HPRCC), hereditary leiomyomatosis RCC (HLRCC), and Birt-Hogg-Dubé (BHD). These hereditary RCC forms are associated with specific, characteristic histologic subtypes. Principally, mutations can be “loss-of-function mutations” in tumor suppressor genes and “gain-of-function-mutations” in proto-oncogenes, which are then called oncogenes.
Research regarding hereditary clear cell RCC has led to the identification of a relevant gene locus on the short arm of chromosome 3. This loss-of-function-mutation led to the assumption of the existence of a tumor suppressor gene, and subsequently, further research led to the identification of the VHL gene. VHL is an hereditary cancer syndrome in which affected individuals have a high risk for the development of tumors in multiple organs, including cerebellar and spinal hemangioblastomas, retinal angiomas, pancreatic neuroendocrine tumors, pheochromocytoma, and bilateral, multifocal clear cell RCC. Loss of VHL also occurs in about 50% of the sporadic clear cell RCCs. VHL loss is strongly linked with activation of the hypoxia-induced pathway, which is discussed further later.
Two hereditary forms of papillary RCC are distinguished: HPRCC and HLRCC. The incidence of these hereditary forms of papillary RCC is low, and almost all cases seen by urologists are sporadic tumors. HPRCC is a rare autosomal dominant syndrome associated with type 1 papillary RCC, which is caused by a gain-of-function mutation of the MET proto-oncogene on chromosome 7q, which encodes a transmembrane receptor (c-Met) that interacts with hepatocyte growth factor (HGF). An early- and a late-onset phenotype of HPRCC have been described. Mutations in the MET proto-oncogene are also observed in a small proportion of patients who have sporadic papillary RCC. HLRCC is caused by a mutation of the fumarate hydratase gene, leading to cutaneous and uterine leiomyomas (50% of the affected women have had a hysterectomy by age 30 years) and an aggressive type 2 papillary RCC. About 50% of the affected patients present with nodal or distant metastases. Accumulation of fumarate leads to activation of the hypoxia-induced pathway, because fumarate is a very potent competitive inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase. Thus, increased expression of HIF-1, HIF-2, and glucose transporter 1 (GLUT-1) have been observed in HLRCC.
The BHD syndrome is a genodermatosis characterized by cutaneous fibrofolliculomas, pulmonary cysts, spontaneous pneumothoraces, and an increased risk for multiple or bilateral RCCs. BHD-associated histologic subtypes comprise chromophobe RCC (33%), hybrid chromophobe-oncocytic RCC (50%), clear cell RCC (9%), and oncocytoma (5%). The BHD gene is located on the short arm of chromosome 17 and has the characteristics of a tumor suppressor gene. Mutation or loss of the somatic allele of the BHD gene was detected in approximately 70% of BHD-associated renal tumors.
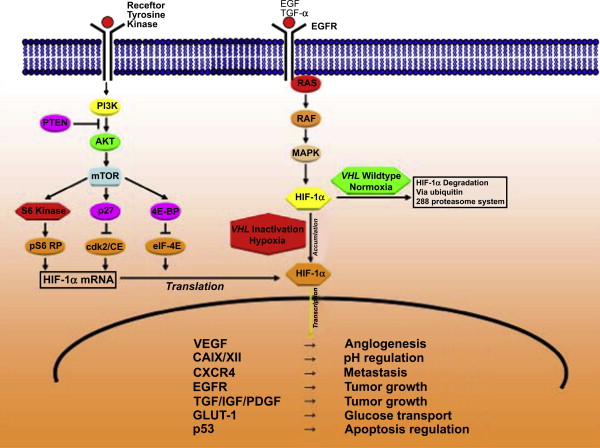
Hypoxia-induced pathway
The hypoxia-inducible pathway plays a key role in regulation of angiogenesis, glucose transport, glycolysis, pH control, epithelial proliferation, cell migration, and apoptosis of RCC, and is responsible for the ability of cancers to adapt to a hypoxic microenvironment. HIF-1 mediates responses to changes in tissue oxygenation by serving as a transcription factor. HIF-1 was identified and purified as a nuclear factor that was induced in hypoxic cells and bound to hypoxia response element (HRE) of the erythropoietin gene. HIF-1 is a heterodimer and consists of a constitutively expressed HIF-1β subunit and a HIF-1α subunit, the latter of which is controlled at the biosynthetic and posttranslational levels. The helix-loop-helix and the PER-ARNT-SIM (PAS) domains mediate heterodimer formation between the two subunits. In addition, HIF-2α can dimerize with HIF-1β and mediate HRE-dependent transcriptional activity. In clear cell RCC, it has been suggested that HIF-2α activation may favor tumor proliferation and growth. A progressive switch to a HIF-2α response may occur during carcinogenesis. This switch may be due to an antisense transcript of HIF-1α, which has been reported to be highly expressed in clear cell RCC. A third member of the HIF-α subunit family, HIF-3α/inhibitory PAS protein, appears to function as an inhibitor and to negatively regulate transcriptional responses to hypoxia.
HIF-1α expression increases with decreasing oxygen concentration, whereas HIF-1β is constitutively expressed. Under normoxic conditions, tumor cells continuously synthesize, ubiquitinate, and degrade HIF-1α protein. In addition, HIF-1α expression is controlled at the posttranslational level by hypoxia through the VHL protein. The first 154 residues of VHL comprise the β domain, which interacts with HIF-1α or HIF-2α. Residues 155 to 213 of VHL comprise the α domain, which interacts with elongin C. An E3 ubiquitin–protein ligase complex consisting of VHL, elongin C, elongin B, CUL2, and RBX1 targets HIF-1α for ubiquitination and proteasomal degradation. Under normoxic conditions, the interaction of VHL with HIF-1α or HIF-2α depends on the hydroxylation of the HIF-α subunit at two proline residues (Pro402, Pro564) by prolyl hydroxylases that utilize oxygen as a substrate. Under hypoxic conditions, the unhydroxylated form of HIF-1α does not bind to VHL; therefore, it is not degraded by the ubiquitin-proteasome. Instead, it migrates into the nucleus, dimerizes with HIF-1β, and binds to HREs within hypoxia-inducible genes and activates their transcription. Another hypoxia-sensing region of HIF-1α is called the carboxy-terminal transactivation domain (CTAD). CTAD binds transcriptional coactivators p300 and CREB-binding protein (CBP) and activates transcription of genes under hypoxic conditions. Hydroxylation of an asparaginyl residue in CTAD by factor-inhibiting HIF-1 during normoxic conditions down-regulates the function of the HIF-α transactivation domain by preventing recruitment of p300 and CBP. In addition to hypoxia, inactivation of the VHL tumor suppressor gene leads to defective ubiquitination of HIF and subsequently to accumulation of HIF-1α, even in the absence of hypoxia. VHL mutation or gene loss occurs in about 50% to 60% of sporadic clear cell RCC, suggesting a major impact of the VHL-HIF axis in clear cell RCC tumorigenesis.
HIF-α dysregulation results in transcriptional activation of downstream hypoxia-inducible genes, which encode for growth and angiogenic factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), transforming growth factor α, and erythropoietin. Genes encoding for enzymes that are involved in glucose uptake and metabolism (such as GLUT-1) and pH regulation (such as carbonic anhydrase IX [CAIX]) and matrix metalloproteinases are also activated by HIF-1 dysregulation. In addition, loss of VHL leads to (1) HIF-1–dependent inhibition of the CDH1 gene, which encodes E-cadherin, responsible for epithelial cell–cell adhesion and maintenance of tissue architecture, and (2) activation of the MET proto-oncogene, which encodes HGF receptor. In addition, loss of VHL leads to up-regulation of the chemokine receptor CXCR4 (also known as fusin), which plays a role in metastatic spread.
Stimuli other than hypoxia and VHL loss can also induce HIF-1 activation and, subsequently, transcription of hypoxia-inducible genes. For example, signaling by way of the HER2/neu or type I insulin-like growth factor (IGF-I) receptor tyrosine kinase induces HIF-1 expression by an oxygen-independent mechanism. IGF-I–induced HIF-1α synthesis depends on the mammalian target of rapamycin (mTOR) and the mitogen-activated protein kinase pathways, further described in the following paragraphs. Other growth factors, such as epidermal growth factor (EGF) and a dysfunctional phosphatase and tensin homolog deleted on chromosome 10 (PTEN) tumor suppressor gene, also increase HIF-1α expression through this signal transduction pathway. In addition to growth factors, prostaglandin E2, thrombin, angiotensin II, 5-hydroxytryptamine, acetylcholine, and some nitric oxide donors can induce HIF-1 activation under normoxic conditions.
Hypoxia-induced pathway
The hypoxia-inducible pathway plays a key role in regulation of angiogenesis, glucose transport, glycolysis, pH control, epithelial proliferation, cell migration, and apoptosis of RCC, and is responsible for the ability of cancers to adapt to a hypoxic microenvironment. HIF-1 mediates responses to changes in tissue oxygenation by serving as a transcription factor. HIF-1 was identified and purified as a nuclear factor that was induced in hypoxic cells and bound to hypoxia response element (HRE) of the erythropoietin gene. HIF-1 is a heterodimer and consists of a constitutively expressed HIF-1β subunit and a HIF-1α subunit, the latter of which is controlled at the biosynthetic and posttranslational levels. The helix-loop-helix and the PER-ARNT-SIM (PAS) domains mediate heterodimer formation between the two subunits. In addition, HIF-2α can dimerize with HIF-1β and mediate HRE-dependent transcriptional activity. In clear cell RCC, it has been suggested that HIF-2α activation may favor tumor proliferation and growth. A progressive switch to a HIF-2α response may occur during carcinogenesis. This switch may be due to an antisense transcript of HIF-1α, which has been reported to be highly expressed in clear cell RCC. A third member of the HIF-α subunit family, HIF-3α/inhibitory PAS protein, appears to function as an inhibitor and to negatively regulate transcriptional responses to hypoxia.
HIF-1α expression increases with decreasing oxygen concentration, whereas HIF-1β is constitutively expressed. Under normoxic conditions, tumor cells continuously synthesize, ubiquitinate, and degrade HIF-1α protein. In addition, HIF-1α expression is controlled at the posttranslational level by hypoxia through the VHL protein. The first 154 residues of VHL comprise the β domain, which interacts with HIF-1α or HIF-2α. Residues 155 to 213 of VHL comprise the α domain, which interacts with elongin C. An E3 ubiquitin–protein ligase complex consisting of VHL, elongin C, elongin B, CUL2, and RBX1 targets HIF-1α for ubiquitination and proteasomal degradation. Under normoxic conditions, the interaction of VHL with HIF-1α or HIF-2α depends on the hydroxylation of the HIF-α subunit at two proline residues (Pro402, Pro564) by prolyl hydroxylases that utilize oxygen as a substrate. Under hypoxic conditions, the unhydroxylated form of HIF-1α does not bind to VHL; therefore, it is not degraded by the ubiquitin-proteasome. Instead, it migrates into the nucleus, dimerizes with HIF-1β, and binds to HREs within hypoxia-inducible genes and activates their transcription. Another hypoxia-sensing region of HIF-1α is called the carboxy-terminal transactivation domain (CTAD). CTAD binds transcriptional coactivators p300 and CREB-binding protein (CBP) and activates transcription of genes under hypoxic conditions. Hydroxylation of an asparaginyl residue in CTAD by factor-inhibiting HIF-1 during normoxic conditions down-regulates the function of the HIF-α transactivation domain by preventing recruitment of p300 and CBP. In addition to hypoxia, inactivation of the VHL tumor suppressor gene leads to defective ubiquitination of HIF and subsequently to accumulation of HIF-1α, even in the absence of hypoxia. VHL mutation or gene loss occurs in about 50% to 60% of sporadic clear cell RCC, suggesting a major impact of the VHL-HIF axis in clear cell RCC tumorigenesis.
HIF-α dysregulation results in transcriptional activation of downstream hypoxia-inducible genes, which encode for growth and angiogenic factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), transforming growth factor α, and erythropoietin. Genes encoding for enzymes that are involved in glucose uptake and metabolism (such as GLUT-1) and pH regulation (such as carbonic anhydrase IX [CAIX]) and matrix metalloproteinases are also activated by HIF-1 dysregulation. In addition, loss of VHL leads to (1) HIF-1–dependent inhibition of the CDH1 gene, which encodes E-cadherin, responsible for epithelial cell–cell adhesion and maintenance of tissue architecture, and (2) activation of the MET proto-oncogene, which encodes HGF receptor. In addition, loss of VHL leads to up-regulation of the chemokine receptor CXCR4 (also known as fusin), which plays a role in metastatic spread.
Stimuli other than hypoxia and VHL loss can also induce HIF-1 activation and, subsequently, transcription of hypoxia-inducible genes. For example, signaling by way of the HER2/neu or type I insulin-like growth factor (IGF-I) receptor tyrosine kinase induces HIF-1 expression by an oxygen-independent mechanism. IGF-I–induced HIF-1α synthesis depends on the mammalian target of rapamycin (mTOR) and the mitogen-activated protein kinase pathways, further described in the following paragraphs. Other growth factors, such as epidermal growth factor (EGF) and a dysfunctional phosphatase and tensin homolog deleted on chromosome 10 (PTEN) tumor suppressor gene, also increase HIF-1α expression through this signal transduction pathway. In addition to growth factors, prostaglandin E2, thrombin, angiotensin II, 5-hydroxytryptamine, acetylcholine, and some nitric oxide donors can induce HIF-1 activation under normoxic conditions.
Mammalian target of rapamycin pathway (phosphatidylinositol 3-kinase/protein kinase B pathway)
The mTOR pathway has a central role in the regulation of cell growth, and increasing evidence suggests its dysregulation in cancer. Receiving input from multiple signals, the pathway stimulates protein synthesis by phosphorylating key translational regulators such as S6 kinase. The mTOR pathway also contributes to many other critical cellular functions, including protein degradation and angiogenesis.
Signaling through the mTOR pathway begins with activation of membrane-bound receptor tyrosine kinases by circulating growth factor ligands. Their activation leads to autophosphorylation of tyrosine residues in the receptor and transphosphorylation of adaptor proteins. Activation of phosphatidylinositol 3-kinase (PI3K) subsequently occurs on binding of the src homology (SH2) domains to specific phosphotyrosine residues on the activated receptor or associated adaptor proteins. This causes the translocation of the enzyme to the membrane and results in activation of the p110 catalytic unit. Subsequently, activated PI3K phosphorylates phosphatidylinositol-4-phosphate to phosphatidylinositol-3,4-biphosphate and phosphatidylinositol-4,5-biphosphate (PIP 2 ) to phosphatidylinositol-3,4,5-triphosphate (PIP 3 ).
Subsequent signaling from PI3K is mainly mediated through the recruitment of serine/threonine kinases Akt and PDK1. Akt is relocated from the cytoplasm to the inner surface of the plasma membrane. On binding to PIP3, Akt undergoes conformation changes that allow the phosphorylation by PDK1 at Thr308 in the activation loop. Subsequent phosphorylation at Ser473 results in full Akt activation, which translocates to the cytosol and nucleus where it phosphorylates its substrates. Akt promotes cell survival by inhibiting the proapoptotic activity of BAD and caspase-9 and by activating several antiapoptotic proteins such as IκB kinase and cyclic AMP response element binding protein. Inhibition of glycogen synthase kinase-3, p21Cip1, and p27Kip1, and decreased proteolytic degradation of cyclin D1 promote cell cycle progression through the G1/S phase.
mTOR is a downstream target of Akt that plays a critical role in the cell cycle progression from the G1 to the S phase. Activation of mTOR by Akt occurs through inactivation of the tuberous sclerosis complex (TSC). Unphosphorylated TSC2 (tuberin) is bound to TSC1 (hamartin) in a complex that blocks mTOR activation. This complex is disrupted when TSC2 is phosphorylated by Akt, relieving the Rheb GAP activity of TSC2 and allowing Rheb to bind ATP. In the presence of ATP, Rheb switches GDP to GTP and subsequently activates mTOR. The major downstream targets of mTOR are the translational components ribosomal S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1). Activation of S6K1 enhances the translation of mRNAs containing the 50-terminal oligopyrimidine tract, whereas phosphorylation of 4E-BP allows the release of eIF4E that facilitates mRNA binding to the 40S ribosomal subunit. Thus, mTOR stimulates the translation of proteins required for the cell cycle progression from the G1 to the S phase.
PI3K signaling is terminated by degradation of PIP3, which can be mediated by two different phosphatases. SH2-containing inositol phosphatase dephosphorylates PIP 3 at position D5 and thereby produces phosphatidylinositol-3,4-biphosphat, whereas PTEN removes the phosphate group at position D3, thereby converting PIP 3 back to PIP 2 . PTEN function is frequently impaired due to deletions or mutations, resulting in constitutive activation of Akt and up-regulation of the mTOR pathway.
Related posts:
 Pathologic Features of Renal Cortical Tumors
Pathologic Features of Renal Cortical Tumors
 Epidemiology, Clinical Staging, and Presentation of Renal Cell Carcinoma
Epidemiology, Clinical Staging, and Presentation of Renal Cell Carcinoma
 Molecular Imaging of Renal Cell Carcinoma
Molecular Imaging of Renal Cell Carcinoma
 Renal Tumor Natural History: the Rationale and Role for Active Surveillance
Renal Tumor Natural History: the Rationale and Role for Active Surveillance
 Choice of Operation for Clinically Localized Renal Tumor
Choice of Operation for Clinically Localized Renal Tumor
 Lymph Node Dissection in the Management of Renal Cell Carcinoma
Lymph Node Dissection in the Management of Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


