Gunter Wolf, Kumar Sharma Diabetic nephropathy (DN) is the leading cause of end-stage renal disease (ESRD) in most Western societies. It can develop in the course of both type 1 and type 2 diabetes and as a consequence of other forms of diabetes mellitus (DM). Type 1 diabetes is an autoimmune disease characterized by antibody-mediated and cell-mediated destruction of pancreatic islets. Type 1 diabetes may occur at any age but is common in childhood, usually presenting before age 30 years. Type 2 diabetes is characterized by a combination of insulin resistance and insulin deficiency. The metabolic syndrome (insulin resistance, visceral obesity, hypertension, hyperuricemia, and dyslipidemia with high triglyceride levels and low amounts of high-density lipoprotein) is often followed by type 2 diabetes. For a long period, insulin resistance is compensated by increased insulin secretion, but a gradual decline in pancreatic β-cell function finally culminates in hyperglycemia, and type 2 diabetic patients require treatment with insulin. Type 2 diabetes was typically a disease of mostly elderly adults, but recently it is increasingly seen in adolescents and even in children. In addition, other types of DM include maturity-onset diabetes of the young, gestational diabetes, and diabetes secondary to various metabolic disorders or the result of corticosteroid treatment. The risk of nephropathy is strongly determined by polygenetic factors. The risk for development of DN is equal in type 1 and type 2 diabetes, and only 30% to 40% of patients with type 1 or type 2 diabetes will ultimately develop nephropathy. The prevalence of nephropathy in diabetic patients varies among different racial and ethnic groups such that it is relatively increased in African Americans, Native Americans, Mexican Americans, Polynesians, Australian aborigines, and urbanized South Asian immigrants in the United Kingdom compared with Caucasians. Although barriers to care seem likely to account for some of these interpopulation differences, genetic factors may also contribute. Familial clustering of DN has been reported in both type 1 and type 2 diabetes and in both Caucasian and non-Caucasian populations. In a type 1 diabetic patient who has a first-degree relative with diabetes and nephropathy, the risk for development of DN is 83%. The frequency is only 17% if there is a first-degree relative with diabetes but without nephropathy.1 In type 2 diabetes, familial clustering has been well documented in Pima Indians,2 and a familial determinant is also suggested by the observation that albumin excretion rates are higher in offspring of type 2 diabetic patients with nephropathy. The risk is particularly high in the offspring if the mother had been hyperglycemic during pregnancy, perhaps because this causes reduced formation of nephrons (“nephron underdosing”) in the offspring,3 as shown in experimental studies.4 Low birth weight and nephron underdosing are also associated with hypertension, metabolic syndrome, and perhaps DN, although for DN the data are somewhat controversial. The hypothesis has been proposed that nephron underdosing5 leads to compensatory glomerular hypertrophy and increased single-nephron glomerular filtration rate (GFR), thus aggravating glomerular injury if a renal insult such as diabetes occurs. There is ongoing research to identify genetic loci for DN susceptibility through genomic screening and candidate gene approaches. However, risk for DN does not show simple mendelian inheritance, and multiple genes are presumably involved. Whole-genome scanning has identified several chromosomal regions that are linked with DN with susceptibility loci, for example, on chromosomes 3q, 7p, and 18q. At present, the pathophysiologic function of such genetic regions is mainly unknown. Gene polymorphisms may also contribute to familial clustering. A study suggested a genetic predisposition to DN caused by a polymorphism in the carnosinase gene, causing accumulation of carnosine with antioxidant properties.6 Several studies also suggested some detrimental effect of the double-deletion (DD) polymorphism of the angiotensin-converting enzyme (ACE) genotype on disease progression,7 although the finding has not been uniformly confirmed.8,9 Environmental factors, especially dietary factors, may also be involved in the pathogenesis of diabetes and DN. One of the strongest risk factors is the intake of soft drinks containing added sugars such as sucrose or high-fructose corn syrup. Added sugars contain fructose, which has been shown to induce features of metabolic syndrome in both humans and laboratory animals, and the mechanism may be mediated in part by the ability of fructose to increase uric acid level. In turn, an elevated serum uric acid level has been found to be a potent predictor for the development of type 2 diabetes as well as DN. The mechanism is linked to the ability of uric acid to induce oxidative stress and endothelial dysfunction. Recent studies suggest that one of the mechanisms by which low birth weight increases the risk for hypertension and diabetes later in life is because low birth weight results in an elevation of uric acid that persists from birth throughout childhood. Hyperfiltration is common in early diabetes but can be corrected with good glycemic control. Increased GFR involves glucose-dependent effects causing afferent arteriolar dilation, mediated by a range of vasoactive mediators, including insulin-like growth factor 1 (IGF-1), transforming growth factor β (TGF-β1), vascular endothelial growth factor (VEGF), nitric oxide (NO), prostaglandins, and glucagon (Fig. 30-1). Over time, development of vascular disease of the afferent arteriole may result in permanent alterations in renal autoregulation that favor glomerular hypertension. Renal injury in DN is caused not only by hemodynamic disturbances (e.g., hyperfiltration, hyperperfusion) but also by disturbed glucose homeostasis, and the two pathways interact. For example, shear stress increases glucose transport into mesangial cells by upregulation of specific glucose transporters. Furthermore, shear stress and mechanical strain resulting from altered glomerular hemodynamics trigger autocrine and paracrine release of cytokines and growth factors in the glomerulus. Diabetic nephropathy is also associated with tubular abnormalities (e.g., increased tubular reabsorption of sodium). Hyperfiltration increases the colloid osmotic pressure in postglomerular capillaries, facilitating reabsorption of sodium in the proximal tubule. In the presence of hyperglycemia, increased proximal sodium reabsorption may also result from increased activity of the glucose-sodium cotransporter. Angiotensin II (Ang II) also appears to have a role, causing hypertrophic proximal tubular growth and increased sodium reabsorption.10 Renal growth is seen early after the onset of diabetes. The size of the kidney may increase by several centimeters. Glomerular enlargement is associated with an increase in the number of mesangial cells and of capillary loops, thus enhancing the filtration surface area. Enlargement of the glomeruli is the result of cellular hypertrophy, whereas the tubular epithelial cells undergo both proliferation and hypertrophy. In animal experiments, avoidance of hyperglycemia prevents renal hypertrophy. Elevated serum glucose levels cause hypertrophy by stimulating growth factors in the kidney, including IGF-1, epidermal growth factor (EGF), platelet-derived growth factor (PDGF), VEGF, TGF-β, and Ang II. The molecular mechanisms involved in glycemia-induced hypertrophy include induction of cell cycle inhibitors such as p27Kip1. TGF-β stimulates protein synthesis (hypertrophy) but prevents cell proliferation and division by induction of cell cycle inhibitors.11,12 It is overexpressed in the glomeruli and the tubulointerstitium both in experimental and in human DN. Glucose as well as glucose-derived advanced glycation end products (AGEs) and Ang II stimulate the production of TGF-β in mesangial cells, podocytes, and tubular epithelial cells. Hyperglycemia also induces the expression of thrombospondin, a potent activator of latent TGF-β. Treatment of diabetic mice with neutralizing anti–TGF-β antibodies attenuated diabetes-related renal hypertrophy and extracellular matrix (ECM) accumulation and preserved renal function, but had minimal influence on proteinuria. Similarly, inhibition of VEGF prevented glomerular hypertrophy in models of DN and also reduced albuminuria.12 The hallmarks of DN are mesangial expansion, nodular diabetic glomerulosclerosis (the acellular Kimmelstiel-Wilson lesion), and diffuse glomerulosclerosis. The early mesangial lesion is characterized by a variable increase in mesangial cell number and size associated with an increased ECM deposition. In later stages, a general loss of mesangial cells occurs. Increasing evidence suggests that local NO deficiency contributes to these histologic lesions, in particular nodule formation. Indeed, endothelial cell nitric oxide synthase (NOS)–deficient mice made diabetic with streptozotocin represent one of the most promising models for DN. Mesangial expansion is mediated by both glucose and glucose-derived AGEs. In experimental models, mesangial changes can largely be prevented by tight glycemic control or the use of AGE inhibitors such as aminoguanidine. The effects are presumably mediated by TGF-β. Inflammatory processes and immune cells are involved in the development and progression of DN.11 Glomerular and interstitial infiltration by monocyte/macrophages and activated T lymphocytes is observed both in human biopsy specimens and in animal models of DN. An inflammatory state is also suggested by the frequent elevation of serum levels of acute-phase proteins and elevated neutrophil counts. Chemokines and their receptors, in particular monocyte chemotactic protein 1 (MCP-1/CCL2), as well as adhesion molecules seem to contribute to this.11,13 Fractalkine/CX3CL1 may function as an arrest chemokine in monocyte/macrophage adhesion before migration into the kidney. T lymphocyte recruitment is the consequence of glomerular and tubulointerstitial upregulation of RANTES/CCL5 structures as well as tubulointerstitial upregulation of inducible protein 10 (IP-10/CXCL10) (Fig. 30-2). Bardoxolone methyl, as an inducer of the KEAP1-Nrf2 pathway, exhibits anti-inflammatory effects and showed initially promising results as a novel therapeutic agent in patients with type 2 diabetes. However, a large Phase III trial was stopped after an interim analysis revealed significantly more adverse effects and increased mortality in the intervention group (see Chapter 31). Widening of the glomerular basement membrane (GBM) is associated with accumulation of type IV collagen and net reduction in negatively charged heparin sulfate. However, heparin sulfate cannot be the key factor underlying proteinuria, because selective reduction of GBM proteoglycans unexpectedly reduced rather than increased proteinuria. This supports the podocyte being more important in the genesis of proteinuria. Biopsies in patients with DN documented a correlation between the degree of proteinuria and the width of podocyte foot processes. The expression of one permeability-controlling protein, nephrin, is abnormally low in DN.12 The transcription of nephrin is suppressed by Ang II and restored by inhibitors of the renin-angiotensin system (RAS). In addition, in DN, apoptosis of podocytes is triggered by various factors, including Ang II and TGF-β, and adhesion of podocytes to the GBM is reduced by AGE-induced suppression of neuropilin-1. Furthermore, migration of podocytes is also attenuated by the reduction of neuropilin-1, thereby preventing surviving podocytes from covering denuded areas of GBM, and thus causing formation of synechiae and development of focal segmental glomerulosclerosis (FSGS). Crosstalk between glomerular endothelial cells and podocytes involves activated protein C (APC). APC formation is regulated by endothelial thrombomodulin and is reduced in diabetic mice.14 In DN, thrombomodulin-dependent APC formation inhibits podocyte apoptosis (Fig. 30-3). In cultured podocytes, adiponectin administration reduced the permeability to albumin and caused podocyte dysfunction. Endothelial cell dysfunction associated with altered fenestrations and glycocalyx may also contribute to enhanced permeability. Because adiponectin levels are low in patients with the metabolic syndrome or type 2 diabetes, lack of adiponectin may further contribute to proteinuria. In advanced stages of DN, high-molecular-weight serum proteins are able to escape across basal membranes with disrupted texture, gaps, and holes (nonselective proteinuria). Tubulointerstitial pathology is seen early in DN and correlates with prognosis.15 The pathogenic mechanisms underlying tubulointerstitial fibrosis and tubular atrophy are similar or identical to those in nondiabetic progressive renal disease (see Chapter 79): release of growth factors, in particular TGF-β, and other cytokines from the glomerulus and direct or indirect effects of proteinuria. Under the influence of mediators such as TGF-β and Ang II, tubular cells change their phenotype and become fibroblast-like.15,16 High glucose concentration and AGEs further stimulate this process. One of the mechanisms by which glucose may promote tubular disease is by conversion through the polyol pathway to fructose, where it is degraded by local fructokinase to induce oxidative stress and local inflammation. Proapoptotic genes are expressed by tubular cells in DN, as promoted by Ang II. Four different cell types may contribute to matrix accumulation along the glomerular and tubular basement membranes and within the interstitial space: glomerular cells, tubular epithelial cells, macrophages/lymphocytes, and fibroblasts/myofibroblasts. In renal fibrosis, the role of matrix-producing myofibroblasts, which contribute to the progression of fibrosis in DN by facilitating deposition of interstitial ECM, is widely accepted; however, their origin remains controversial. Up to 36% of all interstitial fibroblasts is derived from tubular epithelial cells by epithelial-mesenchymal transition (EMT).16 In addition, endothelial cells and pericytes may also contribute to fibroblast-like transformation. EMT is a highly regulated process defined by four key events: (1) loss of epithelial cell adhesion (e.g., E-cadherin adhesion complex), (2) de novo α-smooth muscle actin (α-SMA) expression and actin reorganization, (3) disruption of tubular basement membrane, and (4) enhanced cell migration and invasion of the interstitium (Fig. 30-4). The third and fourth features are difficult to confirm in human disease. Several factors are known to be important in the induction of EMT and subsequently in tubulointerstitial fibrosis. Most important, the enhanced multifunctional cytokine TGF-β1 has been identified as a central mediator of the fibrotic response as a regulator for EMT. TGF-β1 is increased by hyperglycemia in glomeruli and tubulointerstitium of kidney specimens from patients with established DN, induces fibroblasts to synthesize ECM proteins, such as type I, III, and V collagen and fibronectin. We recently demonstrated that EMT in the diabetic kidney depends on the presence of collagen type VIII, a nonfibrillar collagen protein that exists as a heterodimer composed of two distinct polypeptide chains, α1(VIII) and α2(VIII).17 However, recent findings render the contribution of EMT to renal fibrosis controversial (see Chapter 79). Clinical studies showed that even mild anemia (hemoglobin level <12.5 g/dl for men, 11.5 g/dl for women) increases the risk of progression of DN. Treatment of anemia early in renal failure with erythropoietin (EPO) may slow the decline of renal dysfunction in DN. Anemia presumably causes renal hypoxia. Moreover, hypoxia is exacerbated by the progressive hyalinosis of the afferent and efferent arterioles and loss of peritubular capillaries. In experimental models of chronic renal injury, hypoxia is an important factor aggravating interstitial fibrosis, partly by the induction of factors such as TGF-β and VEGF. The transition of tubular epithelial cells into fibroblasts is stimulated by cellular hypoxia.18 The induction of growth factors and cytokines is mediated by hypoxia-inducing factor 1, which can be amplified by Ang II. Evidence of the role of tight glycemic control in retarding the development of DN includes the following: • Studies suggest that with good glycemic control, as reflected by an average hemoglobin A1c (HbA1c) level of 7.0%, only 9% of type 1 diabetic patients will develop ESRD after 25 years, versus the historical prevalence of 40%.19 • Results from the Diabetes Control and Complications Trial (DCCT) showed a remarkable reduction in progression from normoalbuminuria to microalbuminuria and other microvascular complications, specifically retinopathy, in patients with type 1 diabetes with tight glycemic control.20 At least for cardiovascular sequelae, the benefit of early aggressive reduction of glycemia persisted despite later deterioration of the glycemic control.21 • Euglycemia that followed isolated transplantation of the pancreas was associated with a regression of diabetic glomerulosclerosis after 10 years.22 • Evidence from the United Kingdom Prospective Diabetes Study (UKPDS) showed that reducing the HbA1c level by approximately 0.9% in patients with type 2 diabetes reduces the risk for development of microvascular complications, including nephropathy.23 • The ADVANCE study randomly assigned 11,140 patients with type 2 diabetes to either intensive glucose control or standard glucose control. At the end of 5-year follow-up, HbA1c level was 6.5% in the intensive group and 7.3% in the standard therapy group. Intensive glucose control was associated with a significant reduction in renal events and new-onset microalbuminuria. The trial did not show a significant effect of intensive control on major macrovascular events.24 The mechanisms by which hyperglycemia induces DN are complex and may involve not only effects of elevated glucose levels, but also the generation of AGEs and alcohol sugars (polyols) as a result of hyperglycemia. Brownlee25 proposed a unifying mechanism of how hyperglycemia leads to diabetic complications, including DN (Fig. 30-5). An increase in intracellular glucose concentrations stimulates glucose oxidation in the tricarboxylic acid cycle, pushing more electron donors (NADH, FADH2) into the electron transport chain.25,26 When a critical threshold is reached and electrons are donated to molecular oxygen, superoxide is generated. This mitochondrial overproduction of superoxide activates four major pathways that are involved in DN. Although this hypothesis is supported by studies in cell culture, validation in animal models remains inconclusive, partly because of the difficulty in measuring superoxide production accurately in vivo. Many of the adverse effects of hyperglycemia have been attributed to activation of protein kinase C (PKC), a family of serine-threonine kinases that regulate diverse vascular functions. The activity of PKC, especially the membrane-bound form, is increased in the retina, aorta, heart, and glomeruli of diabetic animals. In short-term and long-term studies of diabetic rats, an orally effective PKC-β–selective inhibitor ameliorated glomerular hyperfiltration, albuminuria, and renal TGF-β overexpression as well as ECM accumulation.12,26 PKC-α may represent an additional therapeutic target because albuminuria was virtually absent in diabetic PKC-α knockout mice. Chronic hyperglycemia can lead to nonenzymatic glycation of amino acids and proteins (Maillard or Browning reaction)27 (Fig. 30-6). Over time, these products undergo rearrangement, including cross-linking, to become irreversible AGEs. Both circulating and tissue proteins as well as lipids and nucleic acids may thus be glycated. Although primarily observed in diabetes, AGEs also accumulate in aging and in renal failure.27 The concentration of AGEs is increased in the serum of DN patients. AGEs have also been localized to diabetic glomeruli by immunohistochemistry. AGEs bind to a variety of cell types, including macrophages and mesangial cells, and mediate a variety of cellular actions, including expression of adhesion molecules, cell hypertrophy, ECM synthesis, EMT, and inhibition of NOS. AGEs injected in vivo induce albuminuria and glomerulosclerosis.27 AGEs have profound effects on podocytes, including induction of hypertrophy, followed by apoptosis and suppression of nephrin synthesis. Among several binding sites, the most important is the putative receptor RAGE (receptor for AGE), a pattern recognition receptor. It is present in tubular cells and podocytes. Ang II stimulates upregulation of RAGE on podocytes.28 This effect is mediated by angiotensin-2 (AT2) receptors not blocked by sartanes.28 One of the actions of RAGE is activation of nuclear factor-κB (NF-κB). sRAGE, the soluble extracellular domain of RAGE, acts as a decoy receptor and experimentally ameliorates the renal lesions in diabetes.27 Administration of aminoguanidine, an inhibitor of AGE formation, to diabetic animals reduces AGE deposition, mesangial matrix expansion, and albuminuria, but has inconsistent effects on GBM thickening. Preliminary clinical studies suggest beneficial effects on retinopathy, lipids, and proteinuria. The clinical experience with aminoguanidine has been disappointing and riddled with side effects. Newer, more specific agents have been developed that may be more potent blockers of AGE formation. For example, phenacylthiazolium bromide cleaves covalent, AGE-derived protein cross-links and provides a conceptual basis for the reversal of AGE-mediated tissue damage, which until now has been regarded as irreversible. The polyol pathway involves the conversion of glucose to sorbitol and eventually fructose. The role of polyols in diabetic complications has been assessed with aldose reductase (AR) inhibitors such as sorbinil, tolrestat, and ponalrestat,12 which have shown promise in preventing diabetic cataracts and improving or stabilizing DN. AR inhibitors also blunt hyperfiltration and have a mild effect on reducing albuminuria in both experimental and human diabetes. Overall, however, the experience with AR inhibitors in DN has been disappointing, and treatment was associated with hypersensitivity reactions and liver function abnormalities. More recently, laboratory studies have focused on blocking fructokinase, which is in the distal polyol pathway, with more promising results. However, fructokinase inhibitors have not yet been administered to humans. Although most of the intracellular glucose is metabolized by the glycolytic pathway, some fructose-6-phosphate is diverted into the hexosamine pathway, increasing the concentrations of N-acetylglucosamine. This glucosamine modifies certain transcription factors, such as Sp1 activity, by post-translational O
Pathogenesis, Clinical Manifestations, and Natural History of Diabetic Nephropathy
Definitions
Pathogenesis of Diabetic Nephropathy
Genetic and Environmental Factors
Hemodynamic Changes
Renal Hypertrophy
Mesangial Expansion and Nodule Formation
Inflammation and Diabetic Nephropathy
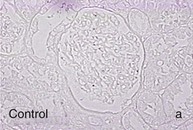
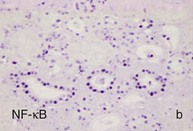
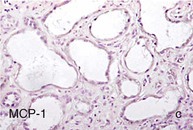
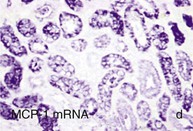
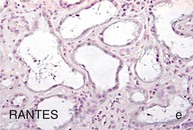
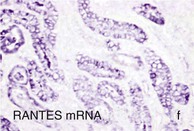
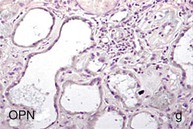
Mechanisms Underlying Proteinuria
Tubulointerstitial Fibrosis and Tubular Atrophy
Hyperglycemia and Diabetic Nephropathy
Role of Glucose Control
Protein Kinase C Pathway
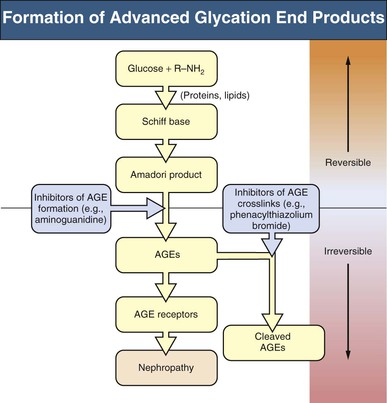
Advanced Glycation End Products Pathway
Polyol Pathway
Hexosamine Pathway
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pathogenesis, Clinical Manifestations, and Natural History of Diabetic Nephropathy
Chapter 30
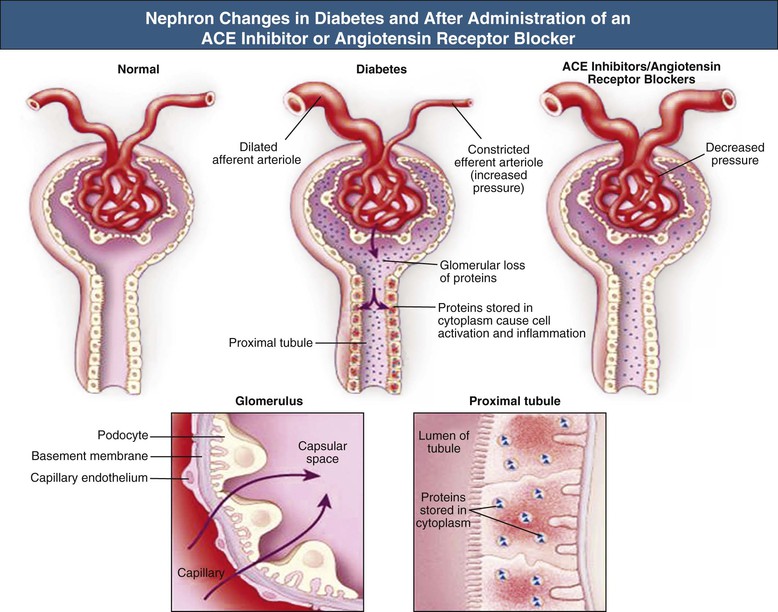
Figure 30-2 Activated nuclear factor (NF)-κB and overexpression of related chemokines in human diabetic nephropathy (DN). Overexpression of chemokines (MCP-1, RANTES), osteopontin (OPN), and activated NF-κB in human DN (a, normal, nondiabetic kidney; b to h, DN) indicating the inflammatory nature of DN. MCP-1, Monocyte chemotactic protein 1; RANTES, regulated on activation, normal T cell expressed and secreted (CCL5); α-SMA, α-smooth muscle actin. (From reference 13.)
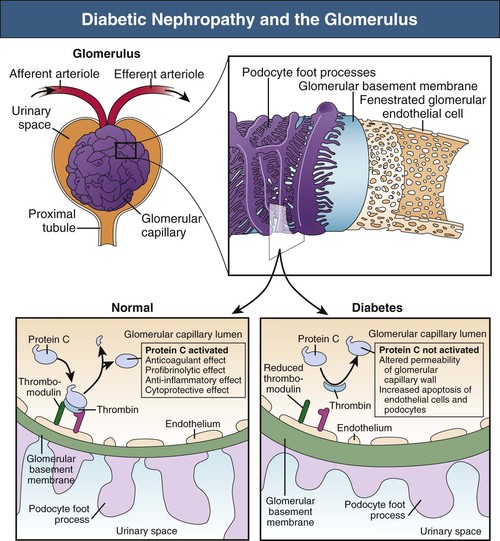
Figure 30-3 Crosstalk between endothelial cells and podocytes involving protein C. Under physiologic conditions, protein C is activated by the binding of thrombin to its receptor, called thrombomodulin, on glomerular endothelial cells. The formed complex catalyzes the conversion of protein C to its catalytically activated form, which has potent anticoagulant, profibrinolytic, anti-inflammatory, and cytoprotective effects. In diabetic nephropathy, the production of activated protein C in the glomerulus is reduced because of suppression of thrombomodulin expression. Decreased functional activity of activated protein C affects the permeability of the glomerular capillary wall and enhances apoptosis of glomerular endothelial cells and podocytes. (From reference 69.)
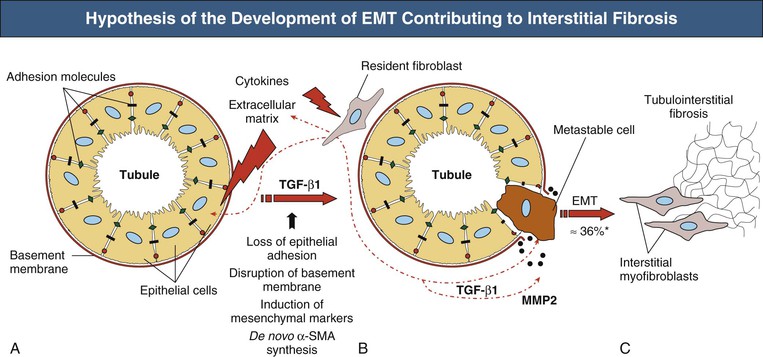
Figure 30-4 Hypothesis of the development of epithelial-mesenchymal transition (EMT) contributing to interstitial fibrosis. Initiated by external stimuli (e.g., cytokines), tubular cells lose their cell-cell contacts (e.g., E-cadherin) (A) and start to express mesenchymal markers (e.g., α-SMA, vimentin) (B). After disruption of tubular basement membrane (by MMP2) metastable cells disengage themselves from cell connective and transdifferentiate to interstitial myofibroblasts that synthesize extracellular matrix and contribute to fibrosis (C). MMP, Matrix metalloproteinase; SMA, smooth muscle actin; TGF, transforming growth factor. *Up to 36% of all interstitial myofibroblasts in diabetic nephropathy are thought to derive from EMT. (Modified from reference 70.)
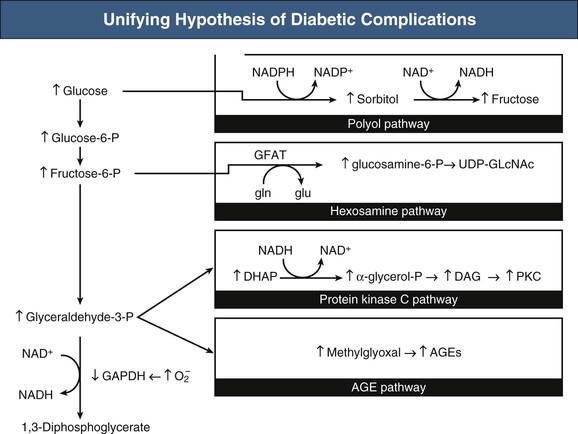
Figure 30-5 Unifying hypothesis of diabetic complications. Mitochondrial overproduction of superoxide activates four major pathways of hyperglycemic damage by inhibiting GAPDH. DHAP, Dihydroxyacetone phosphate; DAG, diacylglycerine (an activator of protein kinase C, PKC, and of triose phosphates to methylglyoxal), the main intracellular advanced glycation end products (AGEs) precursor. Increased flux of fructose-6-phosphate to UDP-N-acetylglucosamine increases modification of proteins by O-linked N-acetylglucosamine (GlcNAc) and increased glucose flux through the polyol pathway consumes nicotinamide adenine dinucleotide phosphate (NADPH) and depletes glutathione (GSH). NAD, Nicotinamide adenine dinucleotide; UDP, uridine diphosphate; GAPDH, glycerinaldehyde-3-phosphate-dehydrogenase; GFAT, glutamine fructose-6-phosphate amidotransferase; gln, glutamine; glu, glutaminic acid; PARP, poly [ADP-ribose] polymerase; P, phosphate. (From reference 26.)