(1)
Al Agouza, Cairo, PO, Egypt
Embryology
The primordial germ cells are first found at the 24th day near the allantois. The germ cells proliferate and migrate to reach the genital ridge, which by the fifth week becomes elevated and thickened. Sex differentiation becomes visible early in the eighth week. Proliferation of oogonia (germ cells) by mitosis continues until approximately the 15th week. By the fourth month, primary follicles appear. In the meantime, the ovary has become recognizable as a discrete organ that “descends” to the level of the pelvic brim and undergoes lateral rotation (Fig. 14.1).
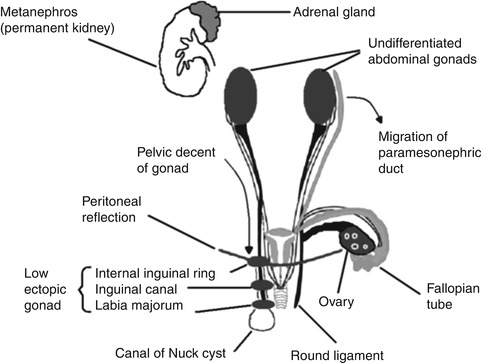
Fig. 14.1
Surgical anatomy of ovary
Gross Anatomy and Relations
The ovaries are ovoid in shape and measure approximately 1.5–3 cm × 1.5–3 cm × 1–2 cm (length × width × thickness) and weigh 2–8 g in adult. The mean volume was 1.06 cm3 (range, 0.7–3.6 cm3) among girls up to 3 months old, 1.05 cm3 (range, 0.2–2.7 cm3) among girls 4–12 months old, and 0.67 cm3 (range, 0.1–1.7 cm3) among girls 13–24 months old. They double in size in pregnancy [1]. The suspensory ligament of the ovary is a peritoneal fold, runs from the side wall of the pelvis to the ovary. The ovarian vessels run in this, crossing over the external iliac vessels. Each ovary is attached to the back of the broad ligament by the mesovarium, which is continuous with its outer coat. A fourth attachment, the ovarian ligament, is a continuation of the round ligament and attaches the ovary to the side of the uterus. Despite all its attachments, the ovary is very mobile, is frequently found behind the uterus in the pouch of Douglas, and has a variable relationship with the uterus:
Anteflexed uterus, lateral or posterolateral
Retroflexed uterus, superolateral
Ovarian anomalies other than the streak ovaries of gonadal dysgenesis are quite rare. Complete absence of an ovary is extremely rare and is usually associated with renal agenesis and absence of the ipsilateral fallopian tube. True ovarian duplication is rarely reported; it occurs in conjunction with duplication of genital ridge and a duplicated Müllerian duct. Excess ovarian tissue near the normal ovarian tissue which has developed from it (and may be connected with it) is classified as an accessory ovary. Lobulation of an ovary is not infrequent and is of little clinical importance. Supernumerary ovaries or the presence of ovarian tissue not connected to the tubes or uterus is very unusual.
14.1 Ovarian Agenesis
Nomenclature
Congenital aplastic ovary
Definition
In ovarian agenesis one or both ovaries are absent and usually accompanies defects of the tubular reproductive organs. Unilateral ovarian agenesis (UOA) and fallopian descent problems are very rare congenital defects.
Confusion exists concerning the terms “agenesis” and “dysgenesis,” and many of the reports in the early literature undoubtedly were examples of streak gonads rather than true agenesis [2].
Incidence
Rarely, one ovary may be absent in an otherwise normal girl; however, unilateral ovarian agenesis is being recognized with increasing frequency in adult females when sexual infantilism calls attention to the related gonadal deficiency; this diagnosis has seldom been made in children before the age of puberty, also few cases had been reported with congenitally absent vagina [3].
Etiology
In some cases, 46,XY gonadal agenesis and 46,XY gonadal dysgenesis may have a common origin, referred to as embryonic testicular regression sequence. Mutations in the gene LHX9, whose murine ortholog causes isolated gonadal agenesis when inactivated, might be responsible for gonadal dysgenesis and agenesis in humans [4].
However, most reported cases of absent ovaries are probably due to antenatal torsion of an otherwise normal fallopian tube and ovary with necrosis and resorption of the adnexal structures.
Unknown environmental factors or genetic predisposition could contribute to this kind of anomaly [3].
Associated findings include agenesis or malformation of the ipsilateral fallopian tube, uterus, round ligament, kidney, and ureter, alone or in combination. Short stature, multiple congenital abnormalities, and high urinary gonadotropins in a young girl should raise the suspicion of absent ovaries.
Diagnosis
In most cases diagnosed after confusion with cases of ovarian dysgenesis or incidentally during routine ultrasound examination for other problems, accurate diagnosis of ovarian aplasia requires a detailed chromosomal analysis, hormonal assay, and MRI examination, and sometimes laparoscopic confirmation is indicated.
Management
Nothing can be done for cases of unilateral absent ovary, but infertility is a normal sequence for cases of bilateral agenesis of the ovaries, and ovarian transplantation for such cases at adolescent to restore ovarian function using ovarian tissue from matched donors, i.e., heterologous transplantation, may be indicated [5].
14.2 Ovarian Dysgenesis
Nomenclature
Turner syndrome, streak gonads
Historical Background
In 1938, Henry Turner first described Turner syndrome, which is one of the most common chromosomal abnormalities in females [6].
Definition
Gonadal dysgenesis generally refers to a condition where gonadal development is abnormal, often only presenting streaks of connective tissue that are often called “streak gonads.” Streak ovaries extend from the lateral pelvic wall to the attachment of the utero-ovarian ligaments. They vary considerably in size; dysgenetic ovaries are characterized by absence of follicular structures and oocytes.
Incidence
Turner syndrome is one of the most common chromosomal abnormalities, occurring in approximately 1 in 2,000 live-born female infants [7].
Etiology
The most common cause of gonadal dysgenesis is Turner syndrome (45X), and most cases have only one normal X chromosome and two-thirds of cases have no other sex chromosome, but in the other third of cases, the girl may have XX gonadal dysgenesis, XY gonadal dysgenesis, or mixed gonadal dysgenesis. So the genotype may be either 45,XO, 46,XX, or 46,XY.
In phenotypic females with a Y chromosome, there is a high risk of the development of a gonadoblastoma, and removal of the gonads is usually indicated. There are also patients of normal height without these abnormalities but with gonadal streaks in which there are two cell lines—one with two normal sex chromosomes and another with only a single X chromosome [8].
This condition will occur if there is an absence of both Müllerian-inhibiting factor and testosterone. The absence of testosterone will result in regression of the Wolffian ducts; normal male internal reproductive tracts will not develop. The absence of Müllerian-inhibiting factor will allow the Müllerian ducts to differentiate into the oviducts and uterus. In sum, this individual will possess female-like internal and external reproductive characteristics, lacking secondary sex characteristics [8].
Other syndromes of ovarian dysgenesis are Swyer syndrome which is also a pure gonadal dysgenesis (46,XY) and Perrault syndrome, XX gonadal dysgenesis with sensorineural hearing loss. Exposure to environmental endocrine disruptors had been incriminated as a predisposing cause, and recently the first gene for Turner syndrome was identified. It is the gene that is responsible for at least part of the short stature in patients with Turner syndrome and is known as the SHOX gene [9].
Clinical Picture
Neonates with streak ovaries often have edema of the hands and feet as a first presenting sign (Fig. 14.2).
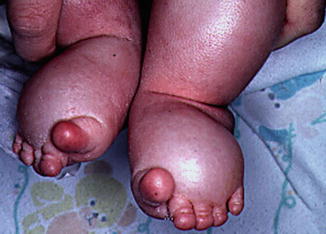
Fig. 14.2
Lymphedema of the feet in an infant is shown. The toes have the characteristic sausage-like appearance
However, many present in adolescence with short stature (<1.5 m); some girls progress through puberty and develop secondary sexual characteristics but enter menopause shortly thereafter.
Turner syndrome is now used to describe girls with sexual infantilism with ovarian streaks, short stature, and two or more of the following: shield chest, obesity, high palate, micrognathia, epicanthal folds, low-set ears, hypoplasia of the nails, osteoporosis, pigmented moles, hypertension, lymphedema, cutis laxa, keloids, coarctation of the aorta, mental retardation, intestinal telangiectasia, and deafness (Figs. 14.3 and 14.4).
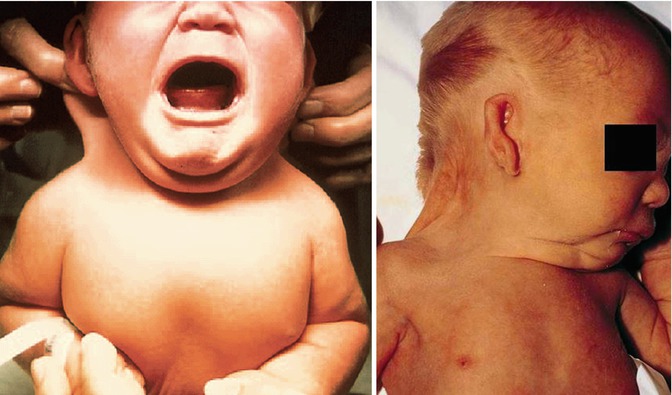
Figs. 14.3 and 14.4
A neonate with webbed neck and low-set ears, typical features for Turner syndrome
Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) are raised, estrogens are low, and non-gonadal endocrine functions are normal.
14.2.1 Diagnosis
Prenatal
On fetal ultrasonography, Turner syndrome is suggested by the presence of a nuchal cystic hygroma, horseshoe kidney, left-sided cardiac anomalies, or nonimmune fetal hydrops, and also diagnosis could be reached by amniocentesis or chorionic villus sampling karyotyping [10].
A standard 30-cell karyotype analysis is required for diagnosis of Turner syndrome, to exclude mosaicism. Diagnosis is confirmed by the presence of a 45,X cell line or a cell line with deletion of the short arm of the X chromosome (Xp deletion) (Fig. 14.5).
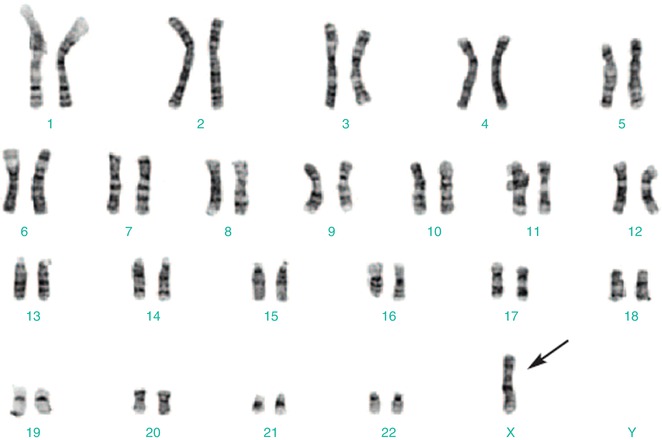
Fig. 14.5
Karyotyping showing a single X chromosome
Management
Patients with Turner syndrome require screening for commonly associated chronic diseases, and early preventive care and treatment are also essential.
Growth Hormone Therapy
In childhood, growth hormone therapy is standard to prevent short stature as an adult. The ideal age for initiating treatment has not been established; taller adult heights occur with the longest treatment durations before the start of puberty [10].
Growth hormone may have long-term favorable effects on lipids, even after it is discontinued.
Sex Hormone Replacement Therapy
Estrogen replacement therapy is usually required, but starting too early or using doses that are too high can compromise adult height.
Estrogen is usually started at age 12–15 years, with low dose can be cycled in a 3-week on, 1-week off regimen after 6–18 months; progestin can be added later. Transdermal estrogens are associated with physiological estrogen levels and may be the preferred treatment, if tolerated [11].
14.3 Ovarian Cyst
14.3.1 Fetal Ovarian Cyst (FOC)
A fetal ovarian cyst refers to an ovarian cysts detected antenatally in a female fetus. They are usually diagnosed in the third trimester.
Historical Background
The first fetal ovarian cyst (FOC) was reported in 1889 as an autopsy finding in a stillborn preterm infant. In 1942 Bulfamonte reported the first case of an ovarian cyst successfully treated during the newborn period [12].
Incidence
Although rare, FOC is the most common cause of intra-abdominal cyst in the female fetal abdomen. From autopsy studies, it is found in up to 30 % of fetuses (this estimate is based on an investigation of stillborns or infants who died within 28 days after birth). With increased use of prenatal ultrasonography, the detection rate for these cysts has increased considerably. Up to 34 % of the fetuses may have antenatally detectable cysts [13].
Etiology
The genesis of FOC is controversial. It may result from fetal exposure to maternal gonadotropins and is observed in fetus whose mothers have increasing levels of human chorionic gonadotropin hormones (hCG) (diabetes mellitus, Rh isoimmunization, toxemia). A precocious FSH peak between 20 and 30 weeks of gestation and abnormal hCG peak due to disorders of theca interna may also be contributory [14].
A fetal ovarian cyst can be of variable size; they tend to be unilateral although bilateral cysts are also rarely seen. It is not thought to change significantly in size over the latter course of the pregnancy (Fig. 14.6).
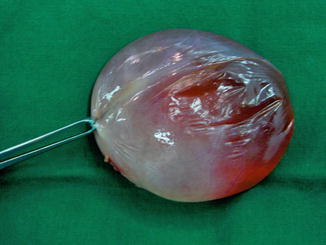
Fig. 14.6
Large simple ovarian cyst
These FOC are benign, functional cysts, which result from enlargement of otherwise normal follicles present in the third trimester and early neonatal period. These cysts have been classified by Nussbaum into simple/uncomplicated and complex/complicated (according to presence of fluid/debris level, clot, septa, echogenic wall) [15].
Associated Anomalies
Prematurity and fetal hypothyroidism are reported; polyhydramnios, which was reported in 5–10 % of the cases of FOC, and other associated anomalies are considered generally rare.
Complications
Common complications include torsion (25–78 %). The risk of ovarian torsion is related more to the length of the pedicle than to the size of the cyst [14] (Fig. 14.7).
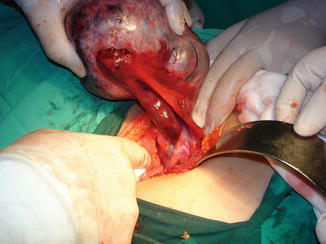
Fig. 14.7
Cystic torsion
Rupture and fetal ascites.
Fallopian tube torsion.
Autoamputation of the ovary.
Compression of adjacent structures.
Antenatal Ultrasound
While it is often difficult to accurately diagnose a fetal ovarian cyst sonographically due to many other cystic lesions having similar appearances, it is typically seen as a well-circumscribed unseptated cyst in the fetal pelvis separate from the fetal bladder, stomach, and gallbladder. It is often anechoic if simple and uncomplicated. The presence of a small daughter cyst is considered a characteristic feature. Due to the relative laxity of supporting ligaments, the cyst can sometimes rise into the upper abdomen (Fig. 14.9). Antenatal MRI could achieve accurate diagnosis in suspicious cases [16].
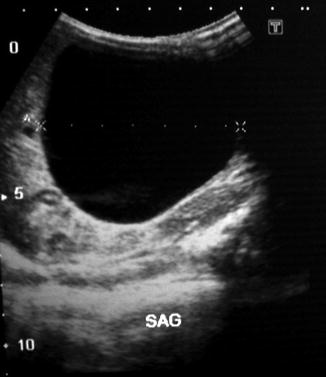
Fig. 14.9
Ultrasound of a large ovarian cyst
Management and Prognosis
Since most of FOC follow a benign course, they have an excellent prognosis especially if the cyst is isolated, unilateral, and unilocular. Spontaneous regression occurs in 25–50 % of cases and is more frequent with smaller cysts. It has been proposed that postdelivery, as the anterior pituitary starts the negative biofeedback mechanism, the abnormal gonadotropin secretion is discontinued, and many of these cysts regress spontaneously although regression may take up to 10 months. As long as simple cysts are small, do not show a trend for rapid growth and remain asymptomatic, they should be monitored by serial ultrasonography during the prenatal period and also postnatally [17]. In the postnatal period, if a cyst is large, does not regress, or increases in size, surgical intervention is recommended to prevent complications such as ovarian torsion and bleeding [18].
14.3.2 Neonatal Ovarian Cysts
Large ovarian cysts may be seen in the neonatal period presenting as an abdominal mass (Fig. 14.10).
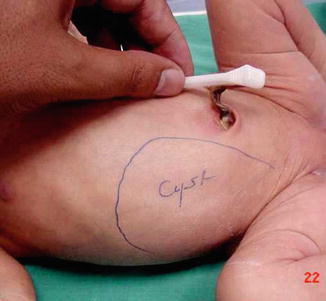
Fig. 14.10
A neonate with large right-sided ovarian cyst
Beyond the newborn period, ovarian cysts in prepubertal children are unusual. Most commonly, however, they are detected prenatally, usually around 26 weeks’ gestation.
Microscopically and histologically, Marshall categorized four types of neonatal ovarian cysts, microscopically distinguished by their lining cells:
1.
Benign cysts of germinal or graafian epithelial origin such as simple cysts, serous cysts, cystomas, cystadenomas, follicular cysts, theca-lutein cysts, and corpus luteum cysts
2.
Benign and malignant granulosa cell tumors
3.
Benign cystic teratomas
4.
Mesonephromas (paraovarian cysts)
Granulosa cell tumors, teratomas, and paraovarian cysts are very uncommon in newborns, and the first general category includes the great majority of neonatal ovarian cysts now reported [19].
The rete ovarii, the ovarian analogue of the rete testis, is a network of anastomosing tubules lined by flat, cuboidal, or columnar nonciliated cells. The rete ovarii rarely gives rise to cysts and to benign and malignant tumors. The cysts occurring in the rete ovarii have been reported only rarely [20].
Investigations
Ultrasonically NOC have a mixed appearance: either simple, containing no internal echoes, or classically containing a fluid/debris level. There is virtually no other abdominal cystic mass in a female infant that produces this fluid/debris level, and a confident diagnosis of an ovarian cyst can usually be made (Fig. 14.11).
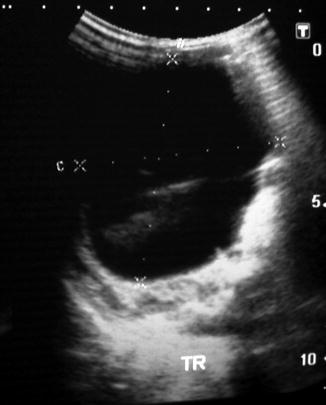
Fig. 14.11
Ultrasound of ovarian cyst showing an intracystic debris
The fluid/debris level is considered to be a sign of preceding ovarian torsion. Such cysts are known as “wandering” tumors and may be found anywhere in the abdomen after becoming detached from the fallopian tube. Rarely the wall or hemorrhagic contents may calcify. The important differential diagnosis is enteric duplications, which can be differentiated by the presence of a well-defined wall and which will not disappear with serial ultrasound examinations. CT scan was used before to confirm the diagnosis in neonates and to distinguish between simple, malignant, or complicated NOC, but due to its potential hazards, it was replaced recently by MRI which is safer and gives a precise delineation of the cyst and its contents (Fig. 14.12).
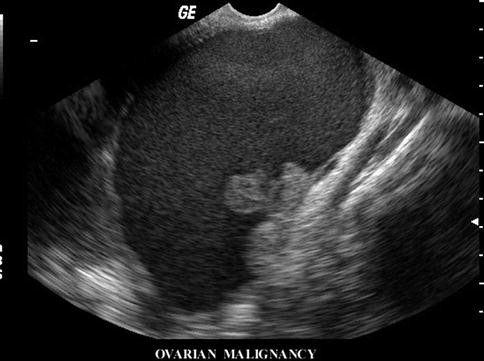
Fig. 14.12
Ultrasound of a large ovarian cyst with internal echoes gives a suspicion of malignant nature of the cyst
Bilateral Neonatal Ovarian Cysts
Both sides of the ovary seem to be affected with equal frequency, and it usually acquires a large size and polyhydramnios is usually manifested early.
Bilateral ovarian cysts are extremely uncommon in the newborn infant, and these cases carry a high risk of removing all normal ovarian tissue (despite attempts to preserve some of each ovary), thereby rendering the girl later on sterile, so great attention should be paid to preserve the ovarian tissue, and only cystectomy is allowed [21] (Figs. 14.13 and 14.14).
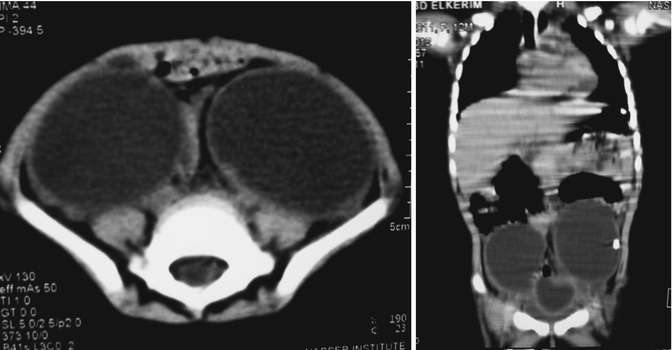
Figs. 14.13 and 14.14
CT scan of bilateral ovarian cysts in coronal and axial sections
Management and Prognosis
The management of neonatal ovarian cysts is conservative, with only serial ultrasound examinations required. The cysts may take a long time to spontaneously involute-up to a year. Rarely surgical intervention needed if they are large (generally over 5 cm) and simple, i.e., transonic and causing pressure effects, for example, respiratory diffi culty; they may be aspirated under ultrasound guidance.
There is no consensus in the modality and timing of the treatment or monitoring of NOC. However, it is certain that surgical treatment is essential in symptomatic, complicated, and torsioned cysts. The major goal of both surgical treatment and noninvasive monitoring by US is optimal ovarian preservation. However, long-term outcome and risk to future fertility are unknown [22].
Small simple cysts under 4 cm in diameter can be kept under observation by serial ultrasonography scan. However, all complicated ovarian cysts and simple cysts >5 cm in diameter should be treated surgically. Minimal access surgery/laparoscopy, being well tolerated by neonates and can be used for aspiration, marsupialization, cystectomy, and oophorectomy or the more conventional open approach can be used.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


