at birth. The continued branching and growth result in the formation of the minor calyces and ever-increasing numbers of collecting ducts (Fig. 1-3).
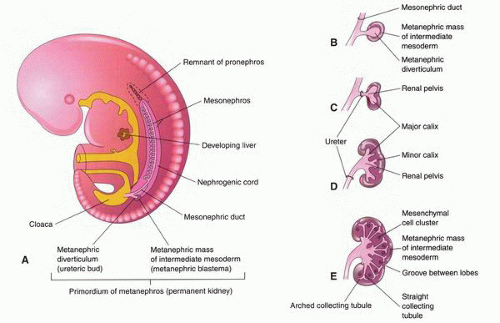 FIGURE 1-1 ▪ Embryologic development of the kidney. A: Sketch of a lateral view of a 5-week embryo showing the primordium of the metanephros. B-E: Sketches showing successive stages in the development of the metanephric diverticulum or ureteric bud (5th to 8th weeks). Note the development of the ureter, renal pelvis, calices, and collecting ducts. (From Moore KL, Presaud TVN. The Developing Human: Clinically Oriented Embryology. 7th ed. Philadelphia, PA: Saunders; 2003, with permission.) |
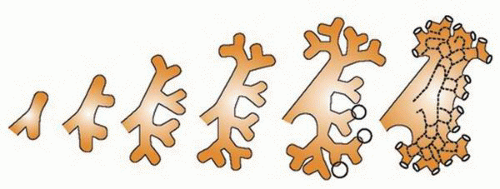 FIGURE 1-2 ▪ Development of the renal pelvis. Diagram showing branching of the ureteral bud. (Reprinted from Mills SE. Histology for Pathologists. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. Modified from Potter EL. Normal and Abnormal Development of the Kidney. Chicago, IL: Year Book; 1972, with permission.) |
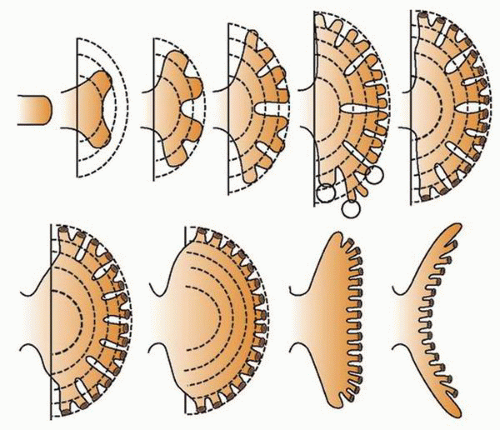 FIGURE 1-3 ▪ Development of renal calyces and papillae. Diagram showing coalescence of the third to fifth generation of branches of the primordial calyx with inward prolapse of the renal papilla. (Reprinted from Mills SE. Histology for Pathologists. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. Modified from Potter EL. Normal and Abnormal Development of the Kidney. Chicago, IL: Year Book; 1972, with permission.) |
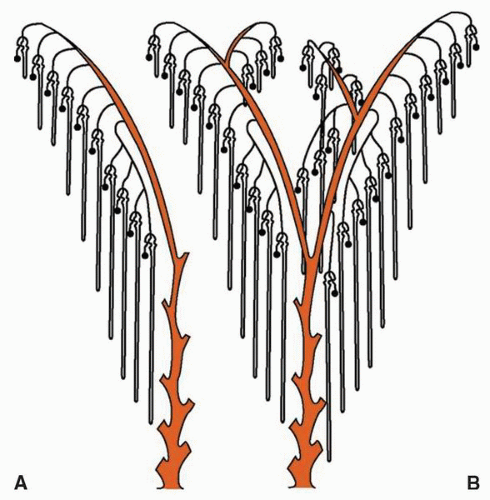 FIGURE 1-4 ▪ Arrangement of nephrons at birth as revealed by microdissection. A: Usual pattern. B: Possible variations. (Reprinted from Mills SE. Histology for Pathologists. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. Modified from Potter EL. Normal and Abnormal Development of the Kidney. Chicago, IL: Year Book; 1972, with permission.) |
parenchyma (Fig. 1-9). Cortical tissue extending from the outer aspect to the renal sinus (columns of Bertin) separates individual medullary pyramids and their draining calyces. The medulla is a darker red brown than the cortex and has a striated appearance. It can be divided into an outer or peripheral zone and an inner zone or papillae. A single unit of a pyramid with its associated cortex is the equivalent of a unipapillary kidney. In humans, the fusion of these individual units results in a multipapillary type of kidney.
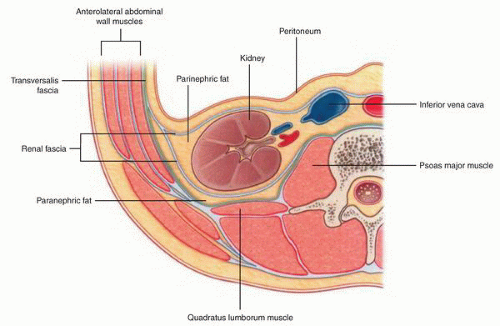 FIGURE 1-5 ▪ Kidney anatomy. Cross section of kidney at the level of renal hilum showing relationship to fat and renal (Gerota) fascia. (From Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia, PA: Churchill Livingstone; 2010:357, with permission.) |
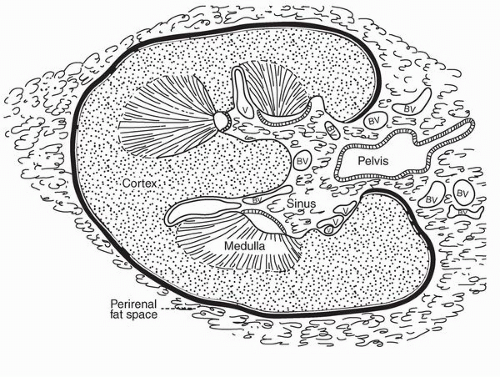 FIGURE 1-6 ▪ Kidney anatomy. Line drawing of cross section of kidney at the level of the renal hilum illustrating the extent of the renal capsule (thick line) and in particular its absence in the renal sinus area. (From Murphy WM, Beckwith JB, Farrow GM. Tumors of the Kidney, Bladder and Related Urinary Structures, AFIP Fascicle. 3rd series. Washington, DC: American Registry of Pathology; 1994, with permission.) |
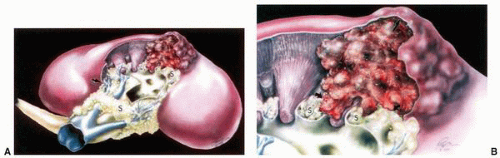 FIGURE 1-7 ▪ Renal sinus and tumor invasion (A,B). Drawing illustrating the propensity of tumors to infiltrate into the renal sinus soft tissue at a location lacking a renal capsule (arrow, B). (From Bonsib SM, Gibson D, Mhoon M, et al. Renal sinus involvement in renal cell carcinomas. Am J Surg Pathol 2000;24:452, with permission.) |
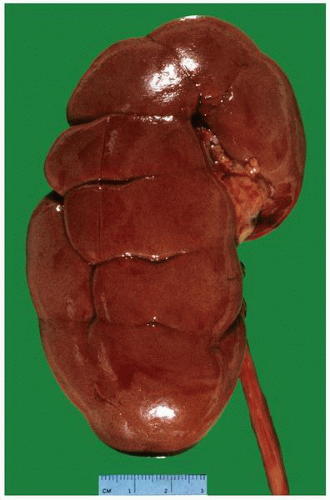 FIGURE 1-8 ▪ Kidney with prominent fetal lobulations. Kidney from a 37-year-old woman with prominent grooves highlighting the normal fetal lobulations that usually disappear with maturity. |
lymphatic system present within the renal capsule drains the outermost portion of the cortex and eventually communicates with the major lymphatic system in the hilar region.
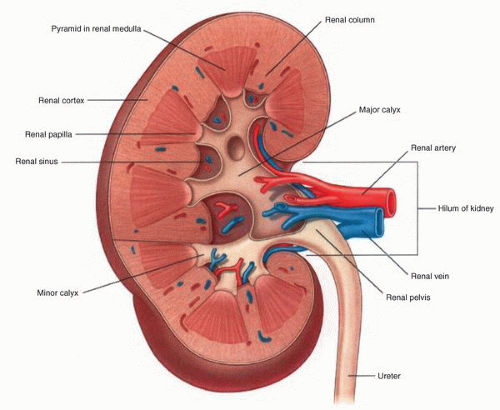 FIGURE 1-9 ▪ Kidney anatomy. Line drawing of the cut surface of the kidney. (From Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia, PA: Churchill Livingstone; 2010:358, with permission.) |
mesangium and fed by the afferent arteriole and drained by an efferent arteriole. The glomerular tuft contains three types of cells: endothelial, mesangial, and epithelial (podocytes) cells. The glomerular visceral epithelial cells (podocytes) transition into the parietal epithelial cells that line Bowman space. These squamous-like cells form a complete layer on the inner surface of Bowman capsule.
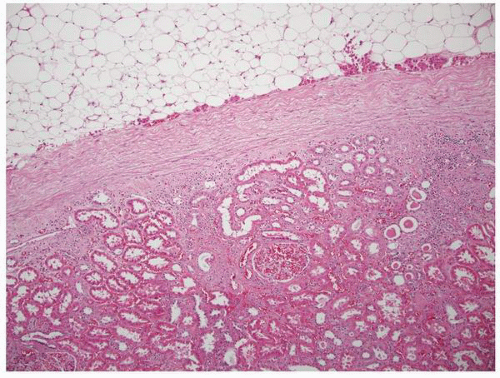 FIGURE 1-10 ▪ Renal capsule. External surface of the kidney illustrating a well-defined fibrous capsule covering the surface of the renal cortex and separating the parenchyma from the perinephric fat. |
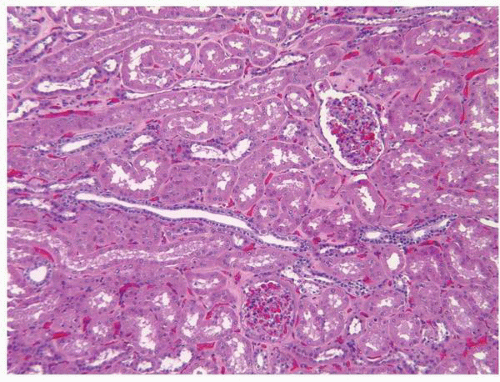 FIGURE 1-11 ▪ Renal cortex. Normal renal cortex containing glomeruli and tubules of the proximal and distal nephron. |
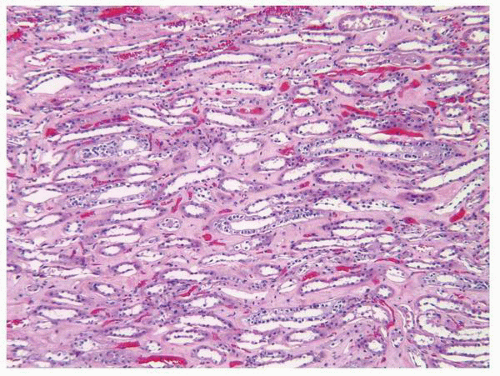 FIGURE 1-12 ▪ Renal medulla. Normal renal medulla with distal tubules and collecting ducts. |
and the intercalated cells. The intercalated cells are largely restricted to the cortical and outer medullary segments of the collecting ducts. By light microscopy, both the principal cells and intercalated cells are cuboidal with pale cytoplasm and centrally located nuclei. The principal cells tend to get taller in the more distal portion of the collecting duct with the cells being columnar in the distal collecting ducts (ducts of Bellini).
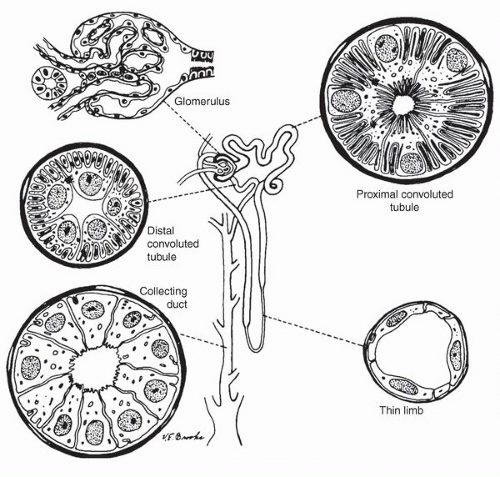 FIGURE 1-13 ▪ Normal histology of the nephron. Line drawing illustrating the various components of the nephron from the glomerulus to the collecting ducts. (From Bennington JL, Kradjian R. Renal Carcinoma. Philadelphia, PA: WB Saunders; 1967, with permission.) |
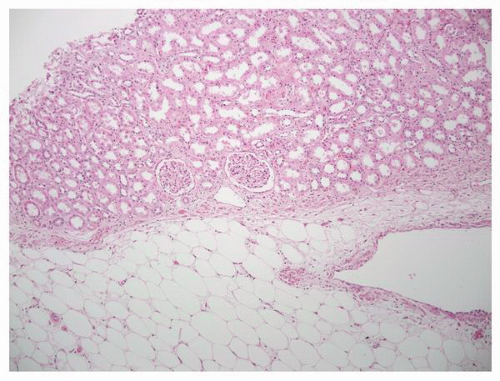 FIGURE 1-14 ▪ Renal sinus. In the area of the renal sinus there is no capsule separating the renal parenchyma from the sinus fat. Note tubular epithelium immediately adjacent to fat cells. |
TABLE 1-1 ▪ SYNDROMES ASSOCIATED WITH RENAL AGENESIS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
orientation. This rotation can be disrupted with the renal pelvis remaining oriented to the anterior or less frequently over rotation results in a more posterior orientation. These abnormalities may have no clinical consequences and might be identified incidentally.22 In others, the abnormal position of the renal pelvis and ureter may result in obstruction and the associated complications including hydronephrosis, obstructive nephropathy, and lithiasis.
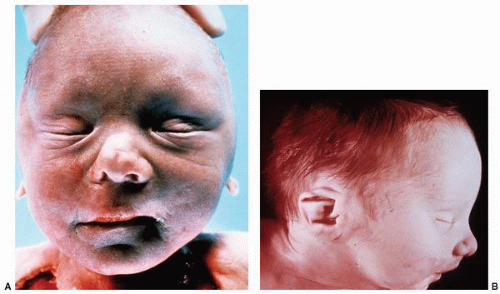 FIGURE 1-15 ▪ Potter syndrome. Infant born with bilateral renal agenesis. Characteristic features of the face include (A) the widely spaced eyes, flattened nose, prominent inner canthic folds, and the receding chin; (B) the lateral view highlights the flattened nose, receding chin, and the low set ears with little cartilage. |
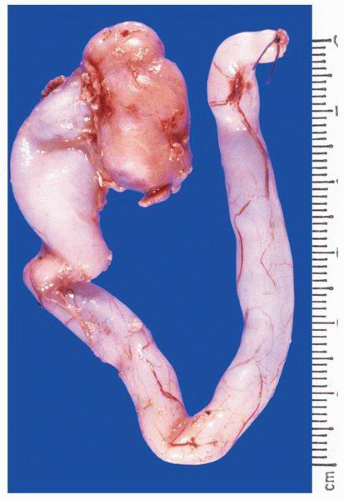 FIGURE 1-16 ▪ Renal hypoplasia. The kidney is small but largely retains a normal reniform shape. |
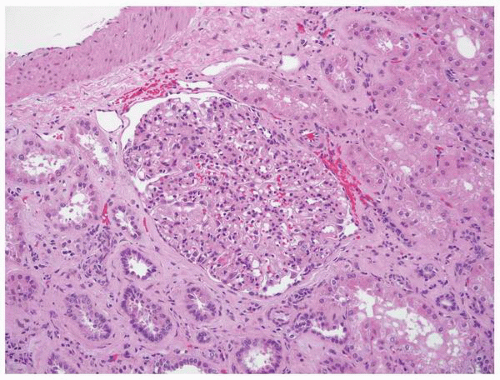 FIGURE 1-17 ▪ Oligomeganephronia. Enlarged glomeruli in the setting of renal hypoplasia. |
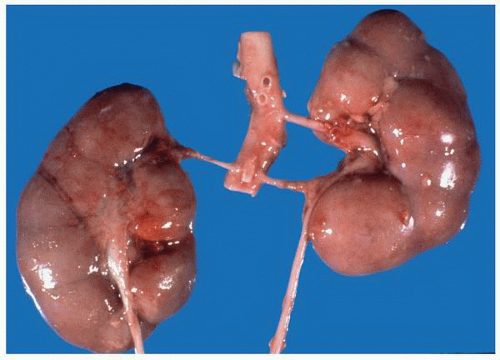 FIGURE 1-18 ▪ Malrotation. The right kidney is abnormally rotated, resulting in a flattened, disc shape. Note also the aberrant renal arteries. |
anomalies, including anomalies of the cardiovascular and central nervous systems. The malposition is typically associated with aberrant vascular and ureteral locations resulting in obstructive phenomena. Many ectopic kidneys are dysplastic. If undetected in childhood, these may present in adulthood with pain, hematuria, recurrent infections, lithiasis, and obstruction. Treatment depends on the specific type of ectopy and its clinical associations.
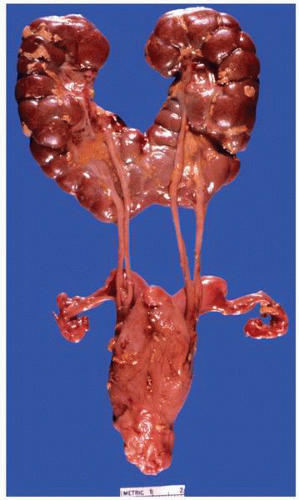 FIGURE 1-19 ▪ Horseshoe kidney. In this example, there is a fairly large isthmus at the lower poles, bilateral bifid ureters, and prominent fetal lobulations. |
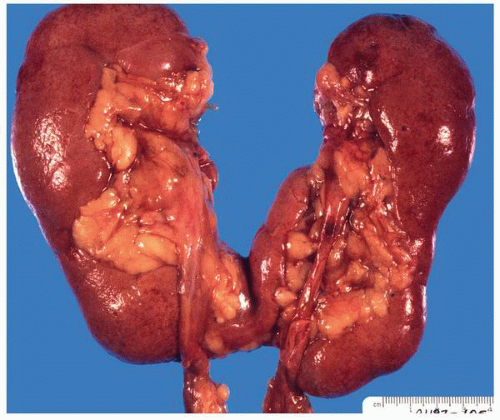 FIGURE 1-20 ▪ Horseshoe kidney. In this case the isthmus joins the lower pole of the right with the lower midportion on the left. |
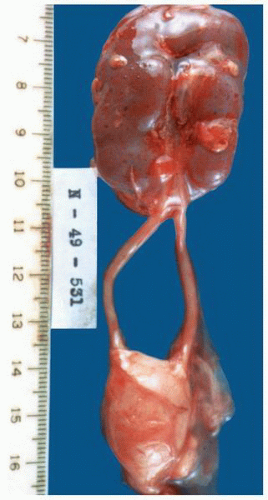 FIGURE 1-21 ▪ Crossed fused ectopia. There is a fused single kidney with two ureters. |
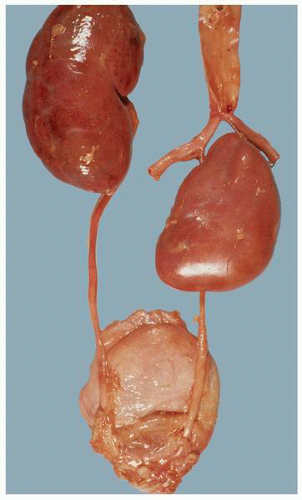 FIGURE 1-22 ▪ Ectopia. The right kidney is located in the pelvis at the bifurcation of the aorta. There is also malrotation with the kidney having a disc-like shape. |
localized to the PKHD1 (polycystic kidney and hepatic disease) gene on chromosome 6.40 This gene codes for the fibrocystin/polyductin protein.41 The protein is localized to the primary cilium within renal epithelial cells of the collecting ducts. It is critical to ciliary function and within the cilium is primarily found in the basal body.42
TABLE 1-2 ▪ CYSTIC DISEASES OF THE KIDNEY | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
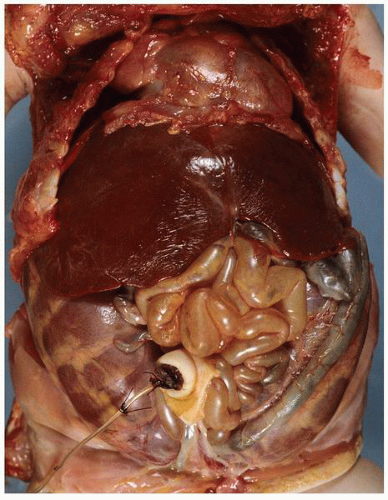 FIGURE 1-23 ▪ Autosomal recessive polycystic kidney disease. In situ photograph of a neonate at autopsy. The markedly enlarged kidneys can be seen filling the abdomen and pelvis. Note the marked displacement of the liver upward resulting in severe compromise of the chest cavity. The lungs were severely hypoplastic. |
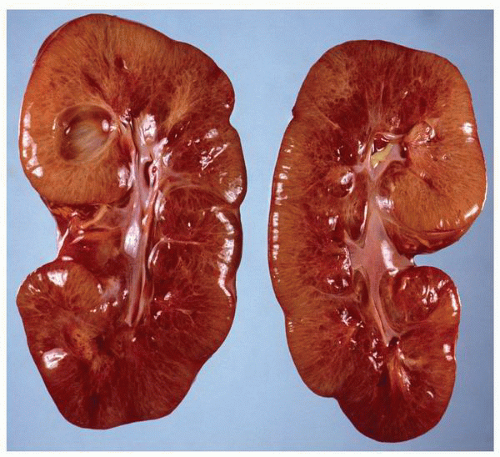 FIGURE 1-24 ▪ Autosomal recessive polycystic kidney disease. Bilateral kidneys at autopsy showing markedly enlarged kidneys with normal reniform appearance. The long tubular cysts can be appreciated in this photograph to result in a radiating pattern outward toward the cortical surface (see Fig. 1-25). |
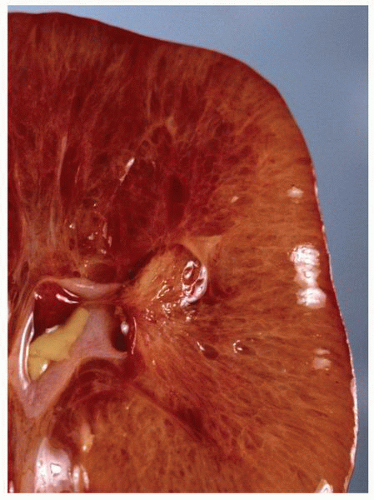 FIGURE 1-25 ▪ Autosomal recessive polycystic kidney disease. Close-up view of the cut surface of the kidney illustrating the elongated fusiform cysts involving the cortex and medulla. |
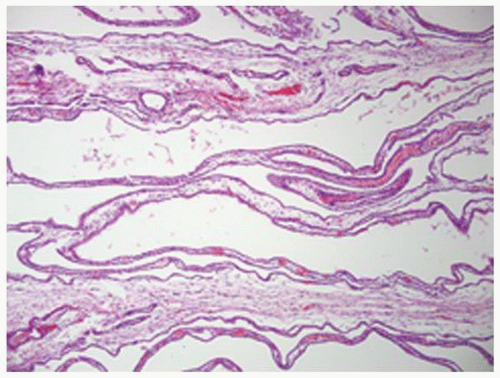 FIGURE 1-26 ▪ Autosomal recessive polycystic kidney disease. The elongated, saccular cysts are lined by a flattened to cuboidal epithelium. |
molecular pathways that are potential targets for therapy67,68 (Fig. 1-27). In transplanted patients, the major causes of death are cardiovascular disease, infection, malignancies, and cerebrovascular causes.55
Table 1-3 ▪ EXTRARENAL MANIFESTATIONS OF AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (APPROXIMATE FREQUENCY) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
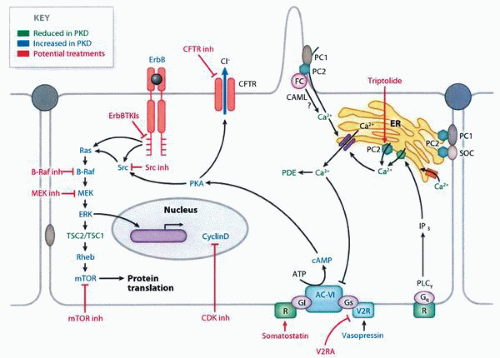 FIGURE 1-27 ▪ Autosomal dominant polycystic kidney disease. Cellular changes with polycystic kidney disease. Components and pathways that are down-regulated and up-regulated are indicated. Potential treatments that target these defective pathways are shown in red. (From Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med 2009;60:325, with permission.) |
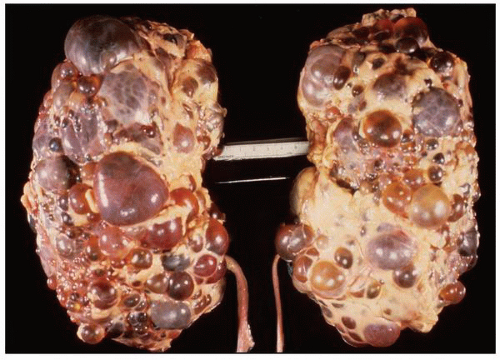 FIGURE 1-28 ▪ Autosomal dominant polycystic kidney disease. Bilateral nephrectomy specimen with markedly enlarged kidneys, normal reniform shape, and the external surface distorted by innumerable cysts of variable size. |
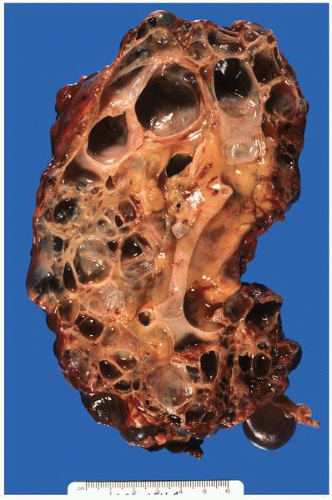 FIGURE 1-29 ▪ Autosomal dominant polycystic kidney disease. Cut surface of a kidney with innumerable cysts replacing the entire kidney. In this 57-year-old patient there is no grossly visible normal parenchyma. |
NPHP3. The NPHP3 gene has been implicated in a small subset of patients with the infantile form.76 The NPHP genes code for a series of proteins known as nephrocystins that are expressed in primary cilia or centrosomes of renal epithelial cells (Fig. 1-33).75,80 It has been suggested that this group of diseases is best classed as “ciliopathies.”81 Cysts presumably develop due to altered signaling pathways resulting in cell polarity and tissue maintenance abnormalities.80
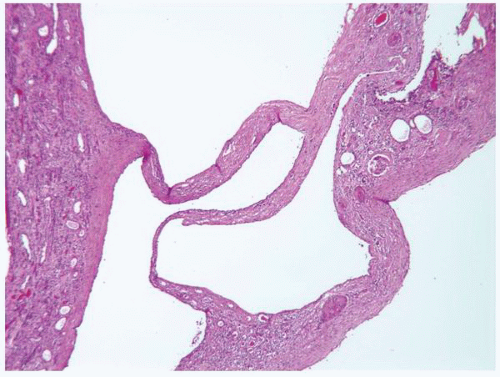 FIGURE 1-30 ▪ Autosomal dominant polycystic kidney disease. Several cysts with varying amounts of stroma containing residual renal parenchyma. In the wider septa residual normal epithelial elements are apparent. |
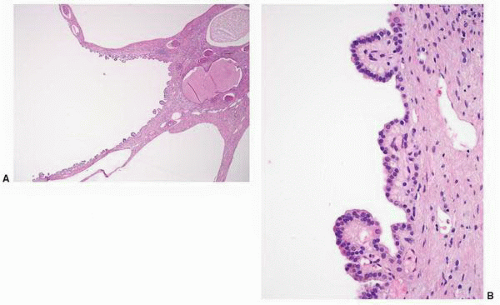 FIGURE 1-31 ▪ A,B: Autosomal dominant polycystic kidney disease. The photomicrograph shows several cysts with varying amounts of stroma and residual parenchyma between them. In one of the cysts, there are multiple small papillae protruding into the lumen. These are a relatively common finding and are covered by cuboidal epithelium without cytologic atypia. |
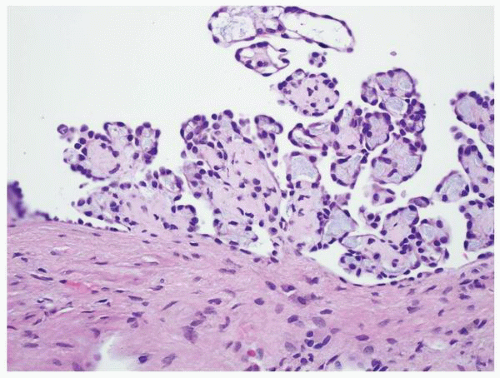 FIGURE 1-32 ▪ Autosomal dominant polycystic kidney disease. Infrequently the papillae become more complex but retain the very bland cytologic features. The nature of these proliferations is unknown, but there is no evidence to suggest they have any clinical significance. |
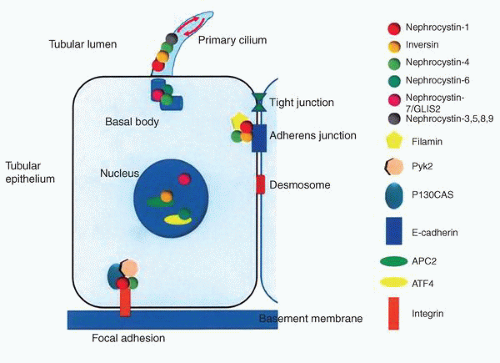 FIGURE 1-33 ▪ Nephronophthisis. Schematic representation of a tubular epithelial cell and the subcellular localization of the nephrocystin proteins. Most of the nephrocystin proteins interact with one another forming a nephrocystin complex. (From Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol 2009;24:2338, with permission.) |
TABLE 1-4 ▪ EXTRARENAL MANIFESTATIONS OF NEPHRONOPHTHISIS | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
fibrosis, tubular basement membrane thickening, atrophic dilated tubules, periglomerular fibrosis, and numerous sclerotic glomeruli resulting in features similar to that found in chronic pyelonephritis. The dilated tubules contain Tamm-Horsfall protein. Ultrastructurally the tubular basement membranes show patchy basement membrane thickening with duplication alternating with areas of splitting and thinning. The basement membrane changes are seen in the juvenile and adolescent but not the infantile forms.75
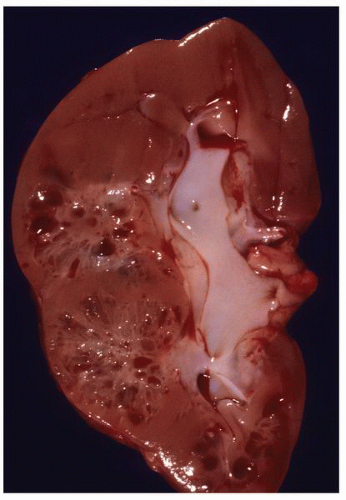 FIGURE 1-34 ▪ Nephronophthisis. Kidney from a patient with juvenile nephronophthisis showing multiple small cysts within the renal medullary pyramids. |
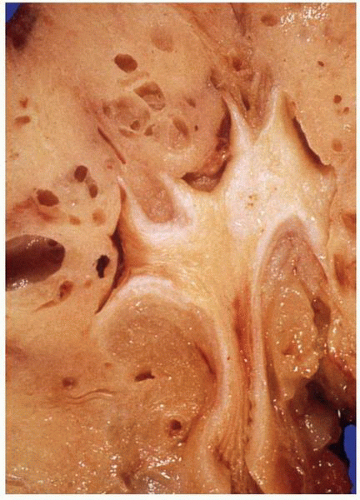 FIGURE 1-35 ▪ Medullary cystic disease. Kidney showing multiple cysts within the renal medullary areas. |
cysts can be multilocular. Tumors are typically multifocal with a variegated appearance (Fig. 1-37). In a study of 33 kidneys from 23 patients with von Hippel-Lindau disease, 190 solid lesions were identified.105
Table 1-5 ▪ EXTRARENAL MANIFESTATIONS OF VON HIPPEL-LINDAU DISEASE | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
clear cell proliferation, and individual abnormal clear cells can be present within tubules (Fig. 1-40). These also have an immunohistochemical profile similar to that of clear cell renal cell carcinoma.
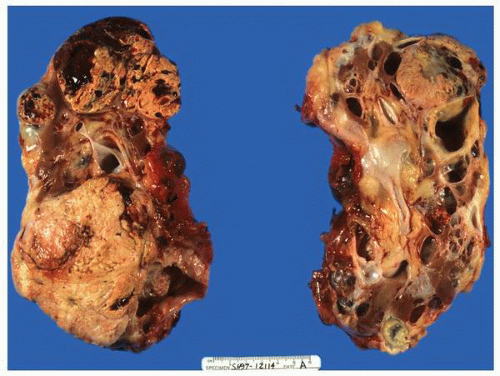 FIGURE 1-36 ▪ von Hippel-Lindau disease. Bilateral nephrectomy from a young woman showing multiple variably sized cysts and multiple clear cell renal cell carcinomas. |
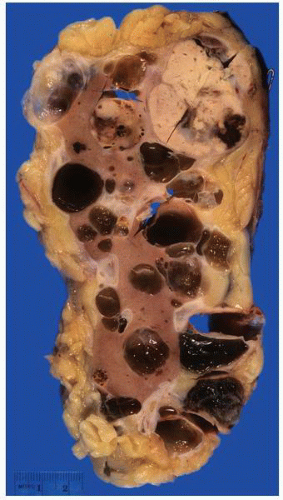 FIGURE 1-37 ▪ von Hippel-Lindau disease. Cut surface of a nephrectomy specimen illustrating multiple cysts with the development of tumors evident within several of the cysts. |
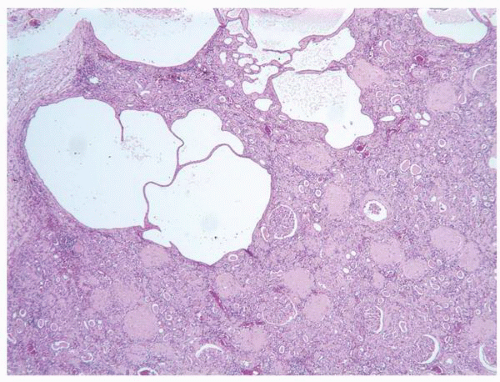 FIGURE 1-38 ▪ von Hippel-Lindau disease. Multiple variably sized cysts lined by flattened epithelium. There are changes secondary to chronic pyelonephritis in the background. |
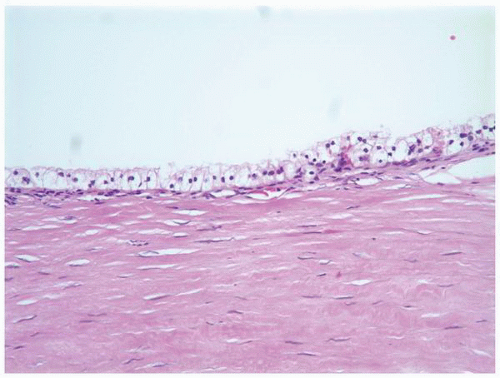 FIGURE 1-39 ▪ von Hippel-Lindau disease. Example of a cyst lined by clear cells with morphologic features typical of the cells of clear cell renal cell carcinoma. |
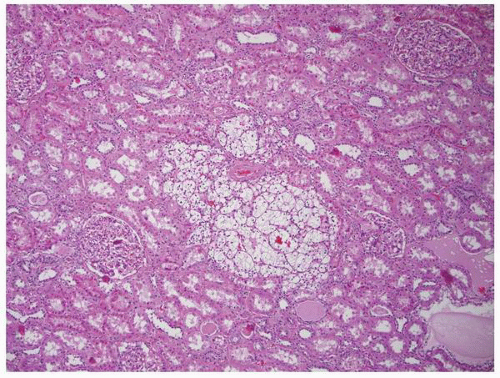 FIGURE 1-40 ▪ von Hippel-Lindau disease. Tiny focus of clear cell renal cell carcinoma. Note the edge of a small cyst in the right lower corner that is lined by clear cells similar to those illustrated in Figure 1-39. |
Table 1-6 ▪ MANIFESTATIONS OF TUBEROUS SCLEROSIS COMPLEX | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
(Fig. 1-41).122,123 In one study, 80% of children with tuberous sclerosis developed renal lesions by age 10 with angiomyolipomas (75%) being more common than cysts (17%).124 Approximately 50% of patients develop renal cysts. The cysts are generally small and can be located in the cortex and medulla. There may only be a few cysts or they can be sufficiently numerous to produce a sponge-like appearance. The tumors range from a few millimeters to several centimeters and have a variable appearance depending on the proportion of the components; hemorrhage is common.
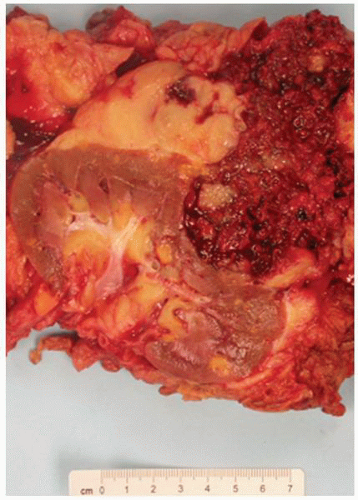 FIGURE 1-41 ▪ Tuberous sclerosis. Nephrectomy specimen with multiple angiomyolipomas (small yellow nodules). The large tumor is an epithelioid angiomyolipoma with rupture and a retroperitoneal hemorrhage. |
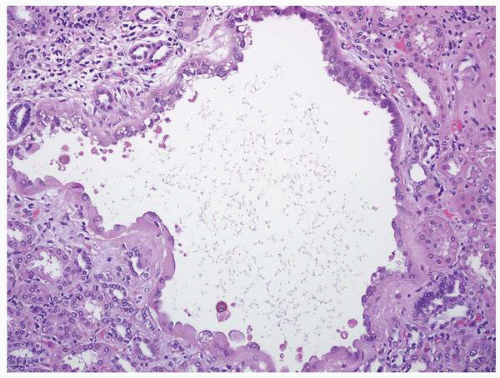 FIGURE 1-42 ▪ Tuberous sclerosis. Cyst lined by cells with moderate to abundant amphophilic cytoplasm. |
been the standard therapy, but conservative treatment with careful monitoring for possible tumor development is an alternative.128 Nephroblastoma and renal cell carcinoma arising in dysplastic kidneys are described.129,130 Intrauterine diagnosis with subsequent treatment prior to birth has been tried with varied success.
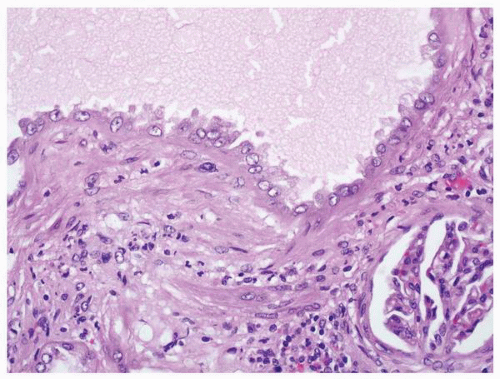 FIGURE 1-43 ▪ Tuberous sclerosis. High-power photomicrograph of the lining epithelium in a cyst. |
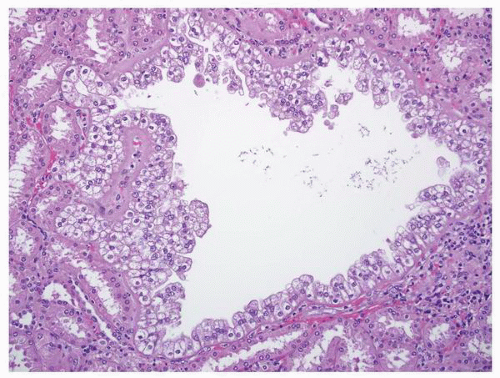 FIGURE 1-44 ▪ Tuberous sclerosis. In this cyst the lining epithelium is pseudostratified and shows cytoplasmic clearing. |
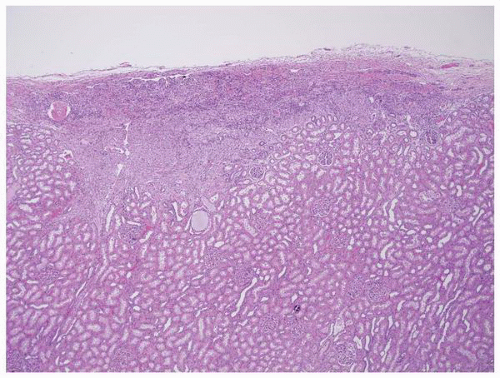 FIGURE 1-45 ▪ Tuberous sclerosis. A small subcapsular angiomyolipoma that is composed entirely of smooth muscle. |
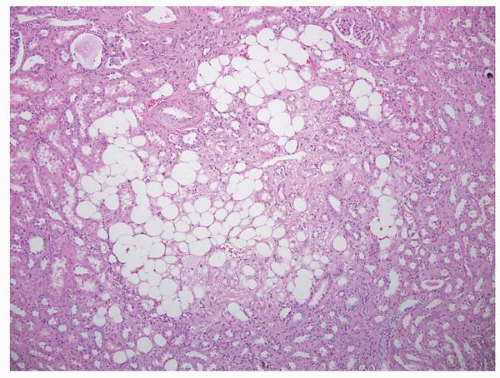 FIGURE 1-46 ▪ Tuberous sclerosis. A small intraparenchymal angiomyolipoma with fat and smooth muscle. |
disease as an entity is reserved for inherited types of the disease. The major form is inherited as an autosomal dominant condition. The subtypes include (i) autosomal dominant glomerulocystic kidney disease related to uromodulin gene (UMOD) mutations, (ii) familial hypoplastic glomerulocystic kidney disease due to mutations in the TCF2 gene (hepatocyte nuclear factor 1β), and (iii) other genetic causes.139 Glomerular cysts occur in a wide range of other conditions including autosomal dominant polycystic kidney disease, autosomal recessive kidney disease, numerous syndromes, and urinary obstruction with or without dysplasia (Table 1-8).139
Table 1-7 ▪ MALFORMATION SYNDROMES ASSOCIATED WITH RENAL DYSPLASIA | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
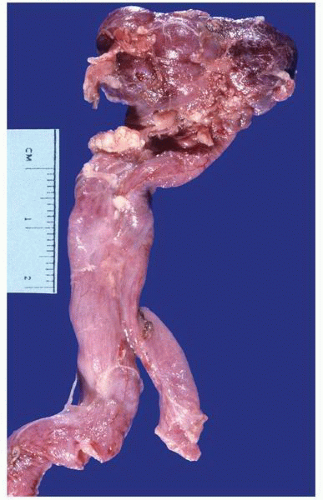 FIGURE 1-47 ▪ Multicystic dysplasia. The kidney consists of a small irregularly, somewhat reniform shaped piece of soft tissue. The cysts are not grossly visible on the surface. There is an associated bifid hydroureter. |
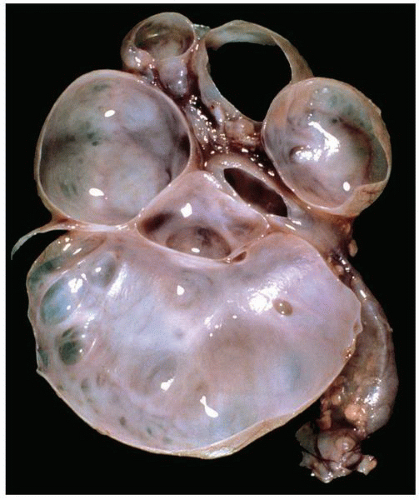 FIGURE 1-48 ▪ Multicystic dysplasia. In this example, the residual kidney is almost entirely cystic with little solid tissue. |
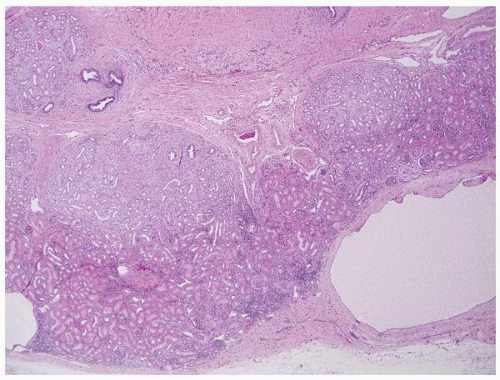 FIGURE 1-49 ▪ Multicystic dysplasia. This is an example of segmental cystic dysplasia with preserved renal parenchyma, cysts, and immature stroma with entrapped tubules. |
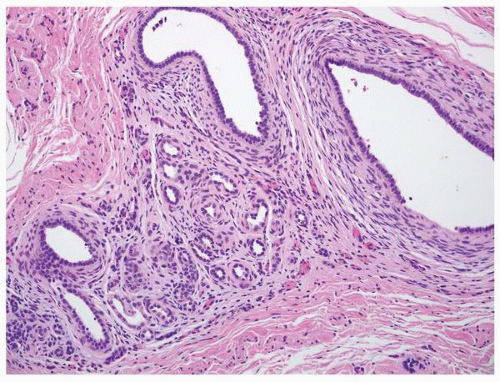 FIGURE 1-50 ▪ Multicystic dysplasia. Small tubules are surrounded by a cuff of immature stroma. |
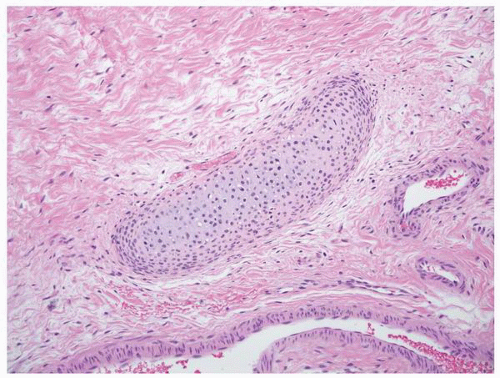 FIGURE 1-51 ▪ Multicystic dysplasia. There is a nest of cartilage within loose fibrous connective tissue. |
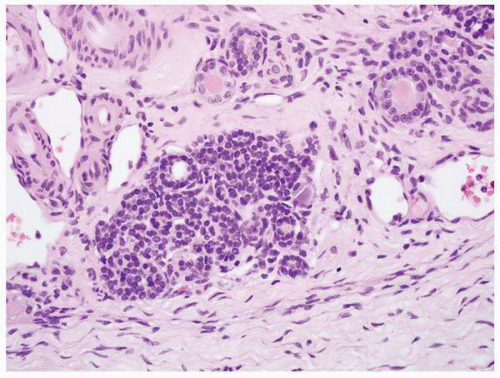 FIGURE 1-52 ▪ Multicystic dysplasia. In this example, there is a nephrogenic rest with adjacent tubules and loose fibrous connective tissue. |
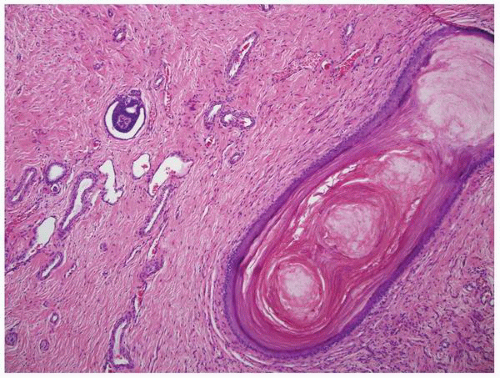 FIGURE 1-53 ▪ Multicystic dysplasia. In this unusual case, the lining of a cyst shows keratinizing squamous metaplasia. |
Table 1-8 ▪ CONDITIONS WITH GLOMERULAR CYSTS | |||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||
and it is seen in association with hemihypertrophy in about 10% of cases.143 The condition is usually asymptomatic and is discovered incidentally in children or in patients in their third or fourth decades presenting with renal lithiasis144,145; the latter occurs in about 50% of patients.146 Diagnosis is usually made by computed tomographic urography.147 These patients rarely progress to chronic renal failure but complications such as recurring urolithiasis, pyelonephritis, and septicemia may cause significant clinical problems.148 Treatment is generally directed at the complications.
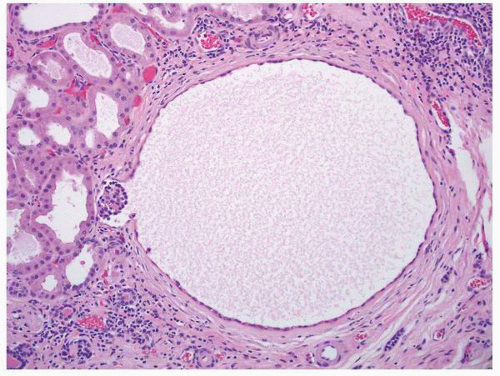 FIGURE 1-54 ▪ Glomerulocystic disease. An example of a glomerular cyst in a case of adult polycystic kidney disease. |
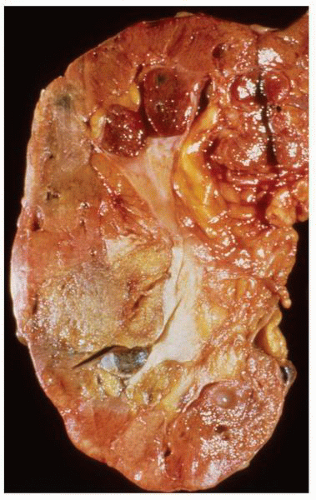 FIGURE 1-55 ▪ Medullary sponge kidney. The kidney shows several small cysts that include several at the corticomedullary junction. |
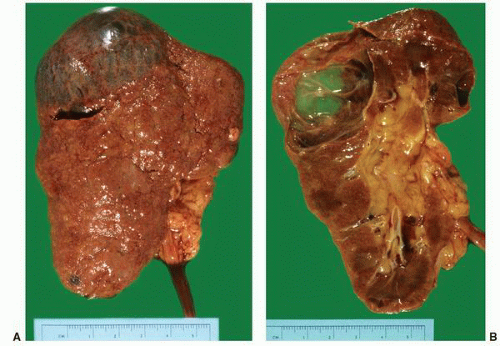 FIGURE 1-56 ▪ Cortical cyst. A large, intact benign cortical cyst can be seen bulging from the external surface of the kidney (A). The opened cyst shows a thin translucent lining (B). |
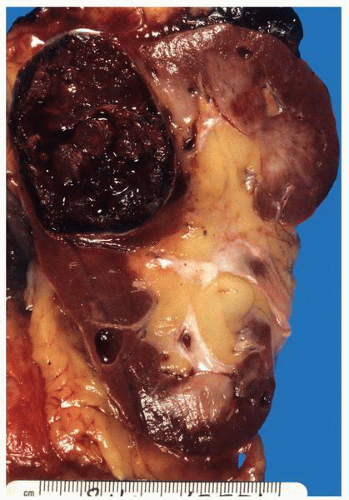 FIGURE 1-57 ▪ Cortical cyst. This is an example of a benign cortical cyst complicated by intracystic hemorrhage. |
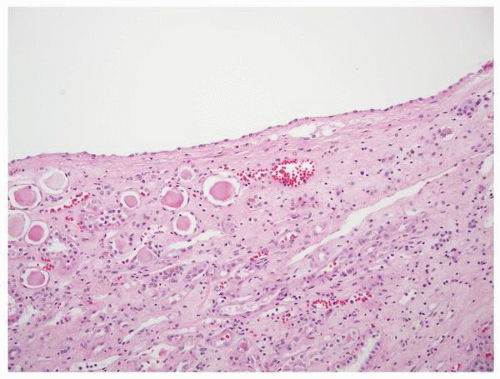 FIGURE 1-58 ▪ Cortical cyst. The lining of an uncomplicated cyst has a flattened epithelium and a thin wall of connective tissue. |
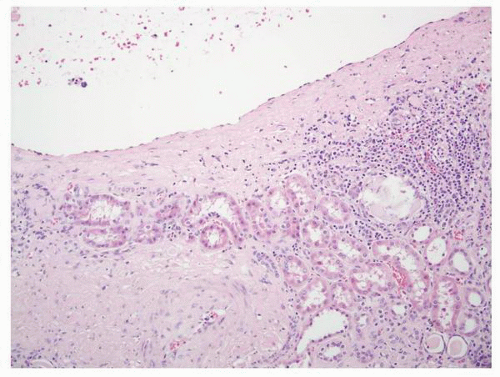 FIGURE 1-59 ▪ Cortical cyst. The wall of this cortical cyst is thickened with dense fibrous tissue, and there is an associated chronic inflammatory infiltrate. |
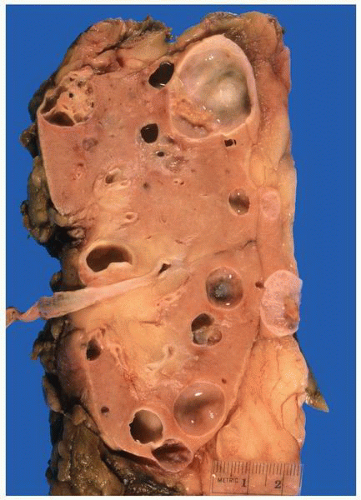 FIGURE 1-60 ▪ Acquired cystic disease. The cut surface of this relatively normal sized kidney shows multiple cysts. The cysts are varied in size and in one a multilocular architecture is present (upper left). Histologically this was a clear cell papillary renal cell carcinoma. |
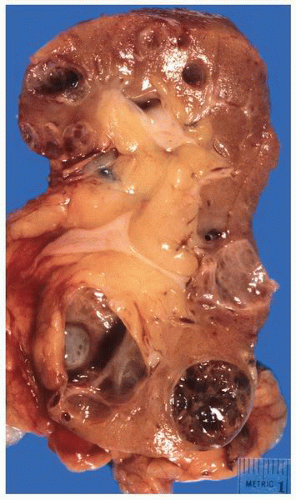 FIGURE 1-61 ▪ Acquired cystic disease. In this example the kidney is small and contains multiple cysts. There is a hemorrhagic tumor arising within a cyst in the lower pole. Histologically the tumor was an acquired cystic disease-associated renal cell carcinoma. |
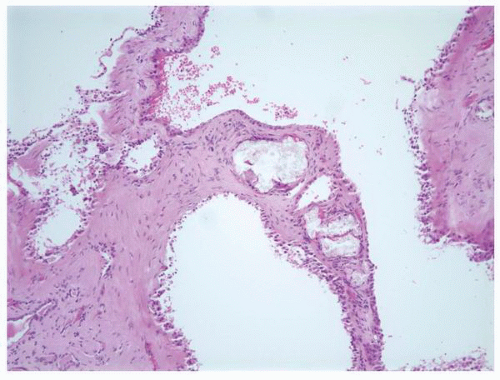 FIGURE 1-62 ▪ Acquired cystic disease. Several cysts in this image are lined by a single- to multilayered cuboidal epithelium. There is fibrotic stroma between the cysts that contains several calcium oxalate crystal deposits. |
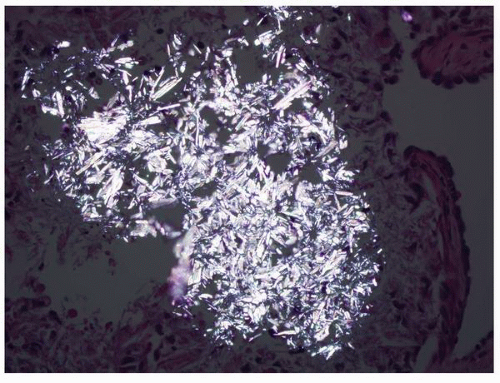 FIGURE 1-63 ▪ Acquired cystic disease. Calcium oxalate crystals as seen with polarized light. |
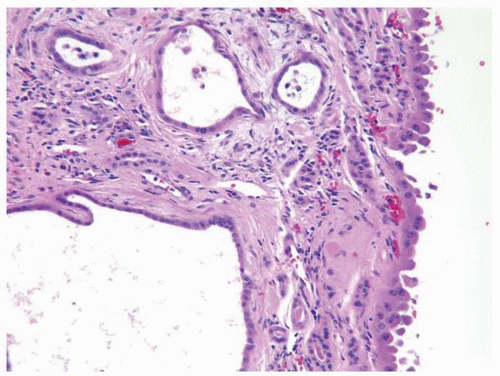 FIGURE 1-64 ▪ Acquired cystic disease. One of the cysts in this photomicrograph is lined by cells with moderate amphophilic cytoplasm and large nuclei that contain prominent nucleoli. |
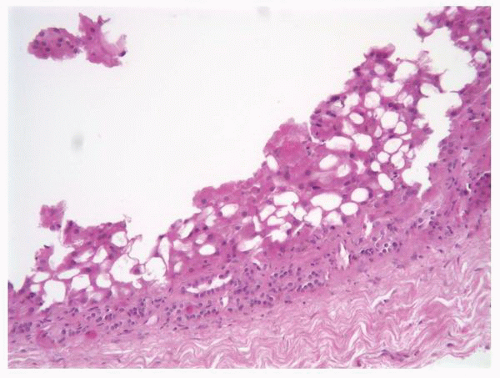 FIGURE 1-65 ▪ Acquired cystic disease. An example of a cyst lined by a pseudostratified epithelium. The cells have abundant eosinophilic cytoplasm and cytoplasmic vacuoles producing a sieve-like appearance. |
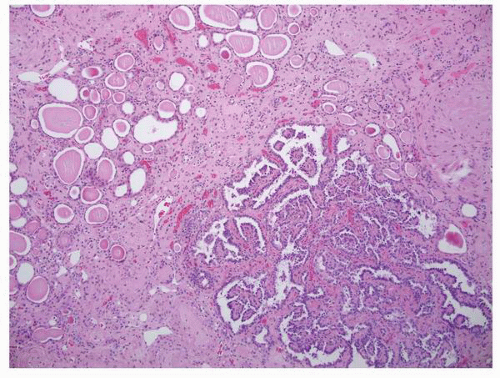 FIGURE 1-66 ▪ Acquired cystic disease. Papillary adenoma developing in a kidney with acquired cystic disease. |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


