TABLE 78.1 Uremic Toxins8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
acetylcholine content and release17 are found in uremic rats. In addition, the basal outflow of GABA and glutamate, but not the K+-stimulated outflow in the hypothalamus, which is measured by microdialysis, is increased in uremic rats. However, the K+-stimulated release of GABA is less sensitive to Ca depletion.18 Whether these changes in neurotransmitters in uremic rats are related to uremic encephalopathy in humans is not clear at present.
| ||||||||||||||||||||||||||
symmetric hypodense lesions in the territories of the posterior circulation, which were reversible after hypertension was controlled. Almost all of them presented with uncontrolled hypertension (frequently > 200 mm Hg systolically). The brain edema is due to the leakage of fluid from hypertension-damaged blood vessels, so called “vasogenic edema.” The edema occurs preferentially in the posterior of the brain, whereas brain edema associated with uremic encephalopathy is diffuse. Because the pathogenesis and treatment of these two diseases are different, it is critical to distinguish them as separate disease entities.
 FIGURE 78.1 Starfruit (Averrhoa carambola), a lethal fruit for the ESRD patient. (From Neto MM, da Costa JA, Garcia-Cairasco N, et al. Intoxication by star fruit (Averrhoa carambola) in 32 uraemic patients: treatment and outcome. Nephrol Dial Transplant. 2003;18:120-125.) |
Silva et al.28 measured most known organic osmoles in the brain after hemodialysis and did not find any significant changes in their concentrations. As for the acidosis hypothesis, it is not clear how intracellular acidosis causes interstitial edema. Although frequently quoted as the pathogenesis of DDS, one may wonder if cerebral acidosis is the consequence, rather than the cause, of brain edema in DDS.
TABLE 78.3 Clinical Parameters of Cases with Dialysis Disequilibrium Syndrome | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the Cardiovascular Health Cognition (CVHS) study, it was found that moderate renal impairment in elderly adults (65 years or older) is associated with a 58% increase in the incidence of vascular dementia, but with no increase in the incidence of pure Alzheimer dementia. The overall incidence of vascular dementia and pure Alzheimer dementia in this cohort is 1.5% and 1.7% per year, respectively.50 In the Northern Manhattan Study (NOMAS), a prospective, community-based cohort of which a subset of stroke-free participants underwent MRIs, CKD stages 3 and 4 are associated with an increase in white matter hyperintensity volume, which is a marker for stroke, cognitive decline, and dementia.51 The increase of white matter lesions in CKD patients is confirmed by Ikram et al.52 More recently, Kobayashi et al.53 demonstrated that CKD also increases silent brain infarcts, and both the prevalence and the number of silent brain infarct increase with declining GFR.53 Furthermore, CKD is also associated with a rapid progression of carotid intima-media thickness in a community study.54
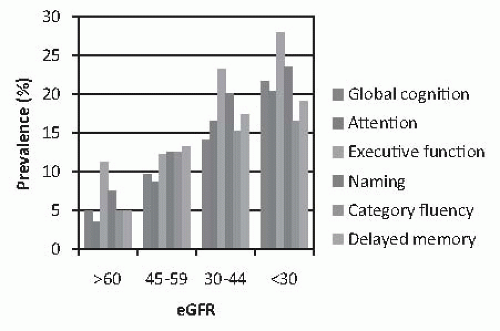 FIGURE 78.2 The unadjusted prevalence of cognitive impairment among 825 older adults (55 years or older) with mild-to-moderate renal insufficiency, according to estimated glomerular filtration rate (eGFR). Data taken from Yaffe K, Ackerson L, Kurella Tamura M, et al. Chronic Renal Insufficiency Cohort Investigators. Chronic kidney disease and cognitive function in older adults: findings from the chronic renal insufficiency cohort cognitive study. J Am Geriatr Soc. 2010;58:338-345. (See Color Plate.) |
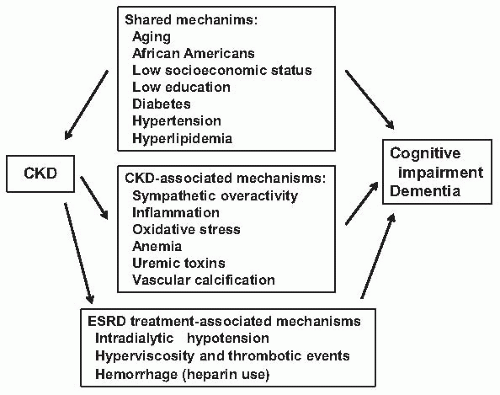 FIGURE 78.3 The proposed mechanisms of cognitive impairment and dementia in end-stage renal disease (ESRD). CKD, chronic kidney disease. (Modified from Kurella Tamura M,Yaffe K. Dementia and cognitive impairment in ESRD: diagnostic and therapeutic strategies. Kidney Int. 2010;79:14-22.) |
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


