or glomerular capillary collapse. These patients are defined as having focal segmental glomerulosclerosis (FSGS), also called focal sclerosis and hyalinosis, and collapsing glomerulopathy, respectively. A spectrum of pathologic findings may be observed. Moreover, some patients, regardless of the underlying pathology, respond to treatment with corticosteroids; others with an apparently identical histologic lesion are resistant to steroid therapy.
TABLE 52.1 Types of Kidney Disease Causing Nephrotic Syndrome in Pediatric and Adult Patients | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 52.2 Classification of Primary Podocytopathies | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
and vascular endothelial (VE)-cadherin.23 The result is a latticework of proteins with openings of approximately 4 × 14 nm.20,24 Therefore, both the GBM and the slit diaphragm contribute to steric influences on macromolecular filtration.
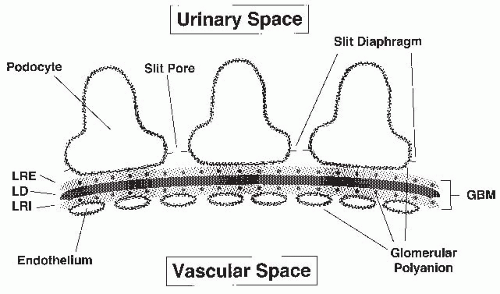 FIGURE 52.1 The glomerular filtration barrier. The distribution of glomerular polyanion in the glomerular basement membrane and on the endothelial and epithelial cell layers is shown. LRE, lamina rara externa; LD, lamina densa; LRI, lamina rara interna; GBM, glomerular basement membrane. |
sufficient capacity that, under physiologic conditions, little intact protein from the filtrate is present in the urine. For example, filtered albumin is subject to lysosomal degradation upon pinocytosis by the proximal tubular cell, with fragments appearing in both the plasma and the urine.42 However, studies of rat kidneys, isolated but perfused in situ with radiolabeled albumin, indicate that some albumin is reabsorbed intact.43 One mechanism of tubular protein reabsorption is demonstrated by its absence in Dent disease, a defect in chloride transport resulting from a mutation in the gene for a renal-specific, voltage-gated chloride channel, CLC-5, leading to hypercalciuric nephrolithiasis.44 Proteinuria in this disease results from disruption of both receptormediated and fluid-phase endocytosis.45 Patients with Dent disease have characteristic urinary losses of retinol-binding protein (RBP) and albumin.46 The failure of protein reabsorption in this lesion has permitted an estimate that the glomerular filtrate contains 22 to 32 mg per liter of albumin, or roughly 3 to 6 g per day in the normal human adult, virtually all of which is reabsorbed under normal conditions. This represents greater than 4% of the total plasma albumin.47
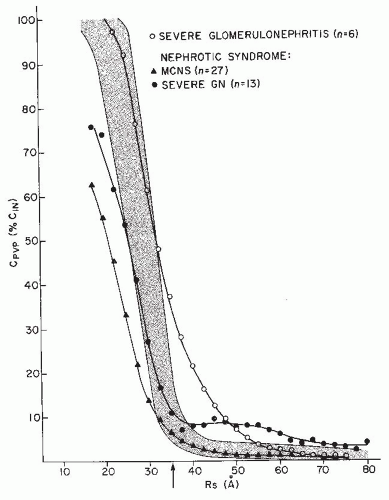 FIGURE 52.2 Permselectivity curves for patients with severe proliferative glomerulonephritis (GN) and for those with nephrotic syndrome secondary either to the minimal change nephropathy or to glomerulonephritis. Normal values are depicted by the shaded area. The arrow indicates the molecular size of albumin. The fractional clearance of larger macromolecules is increased in severe glomerulonephritis. In minimal change nephrotic syndrome (MCNS), the fractional clearance of smaller molecules is decreased. Patients with nephrotic syndrome secondary to glomerulonephritis show a hybrid curve. (Data modified from Robson AM, Cole BR. Pathologic and functional correlations in the glomerulopathies. In: Cummings NB, Michael AF, Wilson CB, eds. Immune Mechanisms in Renal Disease. New York: Plenum; 1982:109, with permission.) |
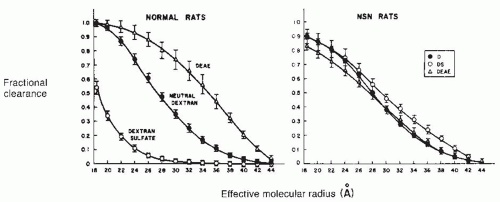 FIGURE 52.3 Clearance of neutral dextran (D), negatively charged dextran sulfate (DS), and positively charged diethylaminoethyl (DEAF) dextran of varying molecular size in normal rats and in those made albuminuric by treatment with nephrotoxic serum (NSN). In normal animals, the clearance of negatively charged dextrans is retarded, and that of cationic dextrans is enhanced, demonstrating charge selectivity by the glomerular filter. In NSN, charge discrimination is lost. (Reprinted from Bohrer MP, Baylis C, Humes HD, et al. Permselectivity of the glomerular capillary wall: facilitated filtration of circulating polycations. J Clin Invest. 1978;61:72; by copyright permission of the American Society for Clinical Investigation.) |
costimulatory signals, is expressed in injured podocytes and binds to cytoskeletal structures, causing foot-process effacement.89,90 It is expressed in the glomeruli of patients with MCN in relapse, but not in remission, and only marginally in patients with FSGS,91 suggesting one mechanism by which podocyte architecture could be disrupted. In several mouse models of proteinuria, circulating levels of soluble urokinase receptor (suPAR) interfere with podocyte adhesive interactions through the adhesion molecules β3-integrin,92 presumably disrupting podocyte architecture through this mechanism as well.
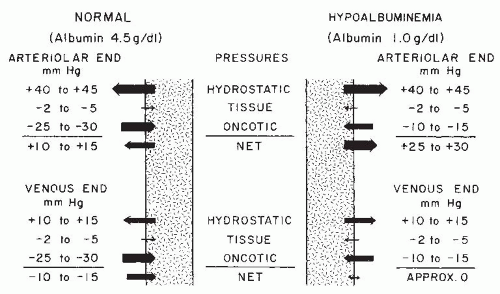 FIGURE 52.4 The forces that govern the movement of fluid across the peripheral capillary wall in healthy persons and in patients with primary nephrotic syndrome. The shaded area represents the lumina of the capillaries. The size and direction of the arrows are in proportion to the magnitude and direction of the force described by that arrow. In minimal change nephrotic syndrome, hypoalbuminemia causes a marked reduction in oncotic pressure. This increases the driving force for fluid out of the arteriolar end of the capillary and decreases the forces available for return of fluid at the venous end. The result is the development of increased amounts of fluid in the interstitial space and the beginning of edema formation. See text for more details. (From Robson AM. Edema and edema forming states. In: Klahr S, ed. The Kidney and Body Fluids in Health and Disease. New York: Plenum; 1984:119, with permission.) |
arterial blood pressure. Pressure is lower in the capillaries (40 to 45 mm Hg) than in the arterial system, but it is markedly higher than tissue pressure, which ranges from 2 to 5 mm Hg. Hydrostatic pressure is opposed by plasma oncotic pressure (the osmotic pressure generated by colloidal solute), which is 25 to 30 mm Hg in healthy individuals. The resulting net force (10 to 15 mm Hg) drives an ultrafiltrate of blood from the capillaries into the interstitial fluid space. By the venous end of the capillary, hydrostatic pressure has been further dissipated (Fig. 52.4) and is exceeded by oncotic pressure, so that there is a net force for the return of fluid into the capillaries. In health, the loss of fluid at the arteriolar end of the capillaries slightly exceeds the amount resorbed at the venous end. The difference is returned to the circulation through the lymphatic system.107
eventually enter into a new steady state in which they no longer accumulate edema and once again demonstrate the ability to excrete a sodium load.111 With this new steady state, sodium and water retention may be so marked and edema accumulation may be so massive that tissue hydrostatic pressure is increased and blood volume is returned to normal. This may explain reports in which nephrotic subjects could be separated into those with high and those with low urine sodium concentrations.120 The high-volume state, whether from massive fluid intake or decreased renal function, represents the overflow pathogenesis of nephrotic edema. It is likely that both underfilling and overflow occur, perhaps at different times in the same patient.
an increase in body weight and a decrease in GFR suggests that prostaglandins may play a role in either the maintenance of GFR or amelioration of edema in nephrotic syndrome. Indomethacin also decreased proteinuria and PRA.144 Response to indomethacin is dependent on concurrent sodium intake. When the agent was given to nephrotic patients on sodium-restricted diets, it resulted in a decrease in GFR. A similar drug regimen for patients with more liberal sodium intake did not affect renal hemodynamics.145
Improvement of renal function occurred in association with diuretic therapy either with or without albumin infusion. In patients who improved with pharmacologic diuresis, the serum creatinine level again rose on return to an edematous state. The authors postulate that glomerular hemodynamics were altered by the presence of intrarenal edema, which occurred concomitantly with peripheral edema.
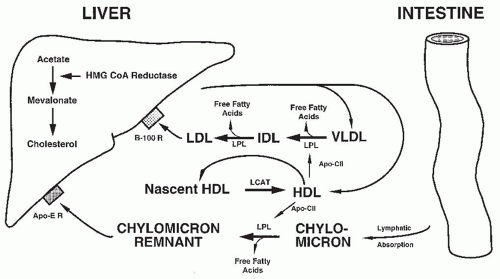 FIGURE 52.5 Normal pathways of lipid metabolism. apo–CII, apolipoprotein C-II; Apo-E R, chylomicron remnant (apo E) receptor; B-100 R, apolipoprotein B-100 (LDL) receptor; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; HMG CoA reductase, 3-hydroxy-3-methylglutaryl coenzyme A reductase; LCAT, lecithin-cholesterol acyltransferase; LDL, low-density lipoprotein; LPL, lipoprotein lipase; VLDL, very-low-density lipoprotein. Figure composed with the assistance of Nader Rifai. |
reductase, mevalonate is produced from acetate in the liver. This in turn is used to make cholesterol, which is incorporated into lipoproteins. The greater the triglyceride content of the lipoprotein, the less dense it is. Dietary fat absorbed from the intestine is formed into chylomicrons by being surrounded with a coat of apolipoprotein (apo) that is critical for transport of the hydrophobic lipid. The triglyceride content of the chylomicron is reduced in the periphery (mainly by the action of lipoprotein lipase [LPL]), and the resulting particle containing apo B-48 and apo E binds to the hepatocyte via a chylomicron remnant receptor. VLDL is synthesized in the liver and metabolized in the periphery through the action of LPL to intermediate-density lipoprotein (IDL), and then to LDL. LDL is bound to apo B-100, which is then taken up by the hepatocyte LDL receptor.189,190 This brings additional cholesterol back to the liver, suppressing HMG CoA reductase activity and decreasing new cholesterol synthesis. The liver also produces HDL, which participates as a transport protein in the catabolism of lower density moieties, being regenerated by lecithin-cholesterol acyltransferase (LCAT). HDL also carries apo C-II, which activates LPL. Abnormalities at any step of metabolism from lipid uptake to the enterohepatic secretion of bile could result in the hyperlipidemia of nephrosis. Likely contributing factors include increased hepatic synthesis of lipoprotein, abnormal transport of lipid through the metabolic pathway, and abnormal catabolism secondary to decreased enzyme activity.
hyperlipidemia. However, lysolecithin, a reaction product that binds to albumin, inhibits LCAT activity in vitro; this feedback mechanism is blocked by the addition of physiologic levels of albumin.214,215 Therefore, the urinary loss of albumin may lead to the inhibition of lipolytic enzyme function.
proteinuria, doses up to 40 mg twice daily caused similar decreases regardless of whether the patients were on corticosteroid therapy. A slight increase was noted in serum HDL concentrations.248 Kinetic studies showed that lovastatin enhances the catabolism of VLDL triglycerides and lowers LDL cholesterol by decreasing input rates for LDLs,249 most likely through the inhibition of LDL-apo B synthesis from VLDL.250 Atorvastatin also is effective in reducing nephrotic hyperlipidemia.251 In children, HMG CoA reductase inhibitors may be effective, but some clinicians have urged caution regarding their use in the very young child, raising the possibility that inhibiting cholesterol synthesis might impair neural myelination. This potential concern needs to be weighed against the more immediate issues of cardiovascular complications and renal disease progression.
thrombin; α1-antiplasmin and α2-macroglobulin inhibit plasmin; and the plasminogen activator inhibitors (PAIs) inhibit plasminogen activators and aPC. Activated protein C, in turn, opposes PAI effects. Many of the components of these pathways are altered in nephrosis. In addition, physical conditions of the nephrotic syndrome, such as venous stasis, hemoconcentration, increased blood viscosity, and possibly the administration of steroids, may also contribute to enhanced blood clotting. These nephrotic effects on coagulation pathways, which are listed in Table 52.3 and discussed in detail elsewhere,255,262,263 are considered here briefly.
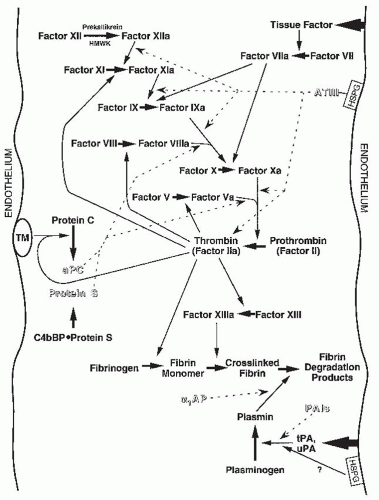 FIGURE 52.6 Interactions among circulating participants in the coagulation cascade. The intrinsic pathway begins in the upper left; the contact system of factor XII, prekallikrein, and high-molecular-weight kininogen (HMWK) that initiates the intrinsic cascade is shown in condensed form. The extrinsic pathway begins in the upper right. The action of factor VIIa on the activation of factor IX has blurred the distinction between these two limbs of the cascade. The common pathway begins with activation of factor × and results in thrombin activity and fibrin cross-linking. Coagulation is counteracted by the fibrinolytic pathway, including plasminogen activators and plasminogen/plasmin. In addition, there are several anticoagulant pathways, most notably inhibition of factors Va and Villa by the inhibitory cofactors protein S and activated protein C (aPC). Alpha-2-macroglobulin, which binds to and inhibits most of the enzymes in this system, is not shown here for the purpose of simplicity. The relationship between heparin sulfate proteoglycans (HSPGs) and plasminogen activator activity shown in the lower right should be regarded as hypothetical for human pathophysiology. Thick solid lines with arrows indicate reactions; thin solid lines with triangular arrows denote catalytic effects; joining lines show cofactors in catalysis. Broken lines with fork-tailed arrows represent inhibitory actions. PAI-1 and PAI-3 bind to aPC; this interaction may cause mutual inhibition of the action of these proteins. α1 AP, α1-antiplasmin; ATIII, antithrombin III; C4bBP, complement factor 4b-binding protein; PAIs, plasminogen activator inhibitors; TM, thrombomodulin; tPA, tissue-type plasminogen activator; uPA, urokinase plasminogen activator. (Figure composed with the assistance of G.A.Soff.) |
TABLE 52.3 Coagulation System Abnormalities in the Nephrotic Syndrome | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
parathyroid hormone levels are increased.328 From these observations, it has been postulated332 that urinary loss of vitamin D complex results in decreased absorption of intestinal calcium, skeletal resistance to parathyroid hormone, and reduced serum calcium levels. In turn, these changes cause increased parathyroid hormone (PTH) production and could result in defective bone mineralization. Although it has not been proved that the changes described cause significant bone disease,333 ongoing steroid therapy may increase the likelihood of clinically significant problems.334,335 In 60 children and adolescents with relapsing nephrotic syndrome, bone mineral density (BMD) was decreased, but whole-body bone mineral content was higher because of an increase in the body-metabolic index.335 In patients with unremitting proteinuria and low 25-OH-D levels, supplementation with oral calcium and 25-OH-D should be considered.336
kilogram initially and increasing, if well tolerated, to 1 g per kilogram per day. The albumin infusion should be preceded by the intravenous infusion of furosemide, 0.5 mg per kilogram. A repeated dose of diuretic is given toward the end of the albumin infusion. Blood pressure should be monitored throughout the albumin infusion to help avoid complications from rapid mobilization of edema fluid into the circulation, although the regimen usually is free from significant side effects when used in children and young adults. Some observations suggest that the administration of albumin may result in more severe glomerular epithelial changes, should raise the oncotic pressure of the tissue space, should delay the response to corticosteroid therapy, and should induce more frequent relapses after remission.347 In view of the potential for complications of albumin infusion,348 this treatment should be reserved for the specific indications of respiratory embarrassment, tissue breakdown, or the need to elicit urine output to confirm the diagnosis of nephrosis.
cholesterol values are maintained.201,251,359,360 Long-term benefits from this therapy have yet to be proved, although anecdotal experience suggests that it may help to reduce proteinuria and maintain GFR,361 as observed in experimental animals (see section on Hyperlipidemia).
twice as often in boys as in girls, whereas it has an equal sex incidence in adults.383 Although no precipitating cause may be apparent in many children, it is not unusual for the development of edema and proteinuria to be preceded by an upper respiratory tract infection, an allergic reaction to an insect sting or other immunogenic stimuli, or the use of certain drugs (Table 52.4).383,384,385,386,387,388,398,400,401,402,403,404,405,406,407,409,410,411,415 In both adult and pediatric patients, malignancies, especially Hodgkin disease, have been associated with the development of MCNS (see section on Nephrotic Syndrome and Malignancies).
TABLE 52.4 Factors Reported to Have Precipitated Minimal Change Nephropathy (MCN) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
enteropathy, can mimic nephrotic syndrome but do not manifest significant proteinuria. The amount of protein in the urine of nephrotic patients can range from less than 1 g to more than 25 g per day. The value for adult patients usually is between 3.5 and 16 g per day; that for children typically is lower than this amount, even when allowances are made for body size419 and averages about 50 mg per kilogram of body weight per day.420 Because urine protein is a function of plasma, and thus of filtrate, protein concentration,421 children with MCNS, who may have serum albumin levels of 1 g per deciliter or less, may occasionally have amounts of urinary protein as low as 100 to 200 mg per day. This finding in patients with low plasma albumin concentrations also reflects the removal of much of the protein from the glomerular filtrate as it traverses the proximal convoluted tubule422 (see previous section on Tubular Handling of Protein in Mechanisms for Proteinuria). As a consequence of proteinuria, the urine specific gravity in nephrosis usually is high, often exceeding 1.035. Exceptions include patients who are not in an edema-accumulating state (see section on Consequences of Proteinuria, earlier in this chapter) and the patient with nephrotic syndrome and renal failure or tubular dysfunction, in whom lower (but not isosthenuric) values of urine specific gravity are found. Physiologic responses of the kidney to the nephrotic state may cause further urine concentration.
are no electron-dense deposits adjacent to the GBM.439 Historically, 65% to 85% of children with primary nephrotic syndrome have this lesion,440 compared to a prevalence of about 30% of primary nephrotic syndrome and 15% of all nephrosis in adults.9
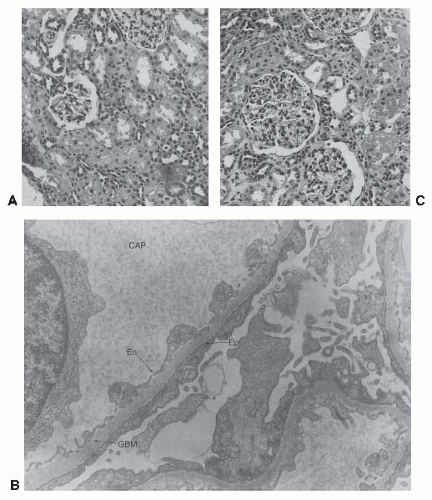 FIGURE 52.7 Findings on renal biopsies from three children with the clinical features of minimal change nephropathy. A: Light microscopy of a patient with MCN. Portions of two glomeruli are shown. Cellularity is normal and the capillary loops are patent. Tubular and interstitial structures are normal in appearance. (Magnification for all light microscopy ×350.) B: Electron microscopy from the same patient. The endothelial cells (En) lining the capillary loop show a normal fenestrated structure; the glomerular basement membrane (GBM) is uniform in thickness and structure. The epithelial cell (Ep) layer shows characteristic fusion of the epithelial foot processes, with the podocytes being in continuous contact with the GBM. Proteinaceous material and a nucleated cell are present in the capillary lumen (CAP). C: Light microscopy in a patient with mesangial hypercellularity. The tubular and glomerular capillary structures are normal, but an increased number of nuclei are present in the mesangial areas of the glomeruli. Immunofluorescent microscopy was negative for immunoglobulins and C3.The patient behaved clinically as one with MCNS. (Histology courtesy of Dr. John M. Kissane.) |
proteinuria, plasma albumin is reduced, which reduces the transmembrane oncotic pressure gradient across the capillary wall and thereby increases PUF In the face of mechanical or hydraulic stress, the capillary wall has three defenses: the mesangial cell, the podocyte, and the GBM. The mesangial cell may undergo proliferation and hypertrophy. Little is known about how the GBM responds to stress. The podocyte cannot proliferate and adapts by elaborating a more complex cytoskeleton to defend the structural integrity of the capillary wall. This occurs, however, at the cost of a reduction in total slit diaphragm width, podocyte effacement, and reduced Kf.
steroid-resistant nephrotic syndrome.10,474 Tip lesion is considered more extensively in the section on Selected Clinical Variants of FSGS.
poor prognosis regardless of renal morphology and (2) patients with a juvenile onset and a generally good response to conventional therapy, provided MCN is found by renal biopsy.
was decreased in MCN patients during the active stage of disease, returning toward normal with remission.531,533,538 Unstimulated secretion of immunoglobulin may be increased,531,537 suggesting spontaneous activation of lymphocytes in MCN. Studies of IgA and IgM synthesis by lymphocytes obtained from patients with nephrotic syndrome of other causes produced more variable results.579 Decreased immunoglobulin production in vitro or in vivo could result from either abnormalities of lymphocytes or the presence of inhibitory agents systemically or on the cell surface. Evidence suggests that both mechanisms may be present in MCN.
TABLE 52.5 Immunologic Abnormalities in Minimal Change Nephropathy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
immune complexes have been documented in some patients with MCN or FSGS,530 not all studies have confirmed this observation529 and the relationship of this finding to mesangial immune complexes remains uncertain. A possible reason for varied results is the use of different assay systems.433 In screening studies that employed liquid-phase and solid-phase C1q binding and Raji cell assays, at least one assay was positive in serum from 11 of 14 adults with MCN, 13 of 27 patients with FSGS, and 26 of 55 patients with membranous nephropathy. Prednisone treatment did not affect the prevalence of circulating immune complexes in this study.602 In another report, 17 of 18 MCN patients had IgG immune complexes that did not bind to Clq; in 7 of 9 patients assessed longitudinally, immune complexes disappeared within 6 weeks of entry into remission.434 This temporal relationship and the absence of glomerular IgG in patients with MCN suggest that circulating immune complexes could be a result rather than a cause of the disease. Although they may not cause the disease, some circulating immune complexes could account for the apparently nonspecific presence of IgM in the mesangium of some patients. In support of this is the finding that neutral, or anionic, large complexes show focal to diffuse mesangial localization.603,604 Complexes containing IgM tend to be large. In contrast, lowavidity, polycationic, and small immune complexes tend to deposit in capillary or mesangiocapillary distribution.
is found in increased concentrations in serum from children with MCN.626 In an animal model, overexpression of IL-13 caused nephrotic-range proteinuria in rats.627
trial of steroids without prior histologic confirmation of the diagnosis. Induction of a complete remission by steroids in such patients is considered to be adequate confirmation of the diagnosis of MCN,440 although other benign conditions may occasionally appear to respond to steroid therapy.640 A histologic diagnosis by renal biopsy is recommended in all patients who present with nephrotic syndrome in the first year of life or after the age of 9 years. Risk-benefit analysis at one point suggested that a biopsy may not be diagnostically superior to a therapeutic trial of steroids, even in adults,641 but the increasing incidence of FSGS in nephrotic adults has led most clinicians to perform a biopsy at the time of presentation. All patients who are steroid-resistant (see Table 52.7 for some commonly used descriptors of nephrotic patients and their responses) should undergo biopsy, although the timing of this decision remains uncertain. Although steroid resistance in children usually is defined by 8 weeks of nonresponse, many clinicians recognize that most children who respond do so by 4 weeks. Therefore, it may be appropriate to perform a biopsy after 4 weeks or less, because those who have not responded by then are more likely to have a different disease that might require an alternative treatment. Other pediatric patients who may require a biopsy include those who have frequent relapses, or who are candidates for therapy with immunosuppressive drugs. Some experts have suggested that any steroid-dependent pediatric patients who continue to be steroid sensitive are, statistically, so likely to have MCN416,461 that biopsy is not indicated. Alternatively, response to steroids may itself be a more important determinant of disease process and outcome than is histopathology.642
TABLE 52.6 Features Suggesting that Nephrotic Syndrome Is Caused by a Disease Other than Minimal Change Nephropathy (MCN) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||









