Nephrogenic and Central Diabetes Insipidus
Daniel G. Bichet
Diabetes insipidus is a disorder characterized by the excretion of abnormally large volumes (greater than 30 mL per kilogram of body weight per day for an adult patient) of dilute urine (less than 250 mmol per kilogram). This definition excludes osmotic diuresis, which occurs when excess solute is being excreted (e.g., glucose in the polyuria of diabetes mellitus). Other agents that produce osmotic diuresis are mannitol, urea, glycerol, contrast media, and loop diuretics. Osmotic diuresis should be considered when solute excretion exceeds 60 mmol per hour. Four basic defects can be involved. The most common, a deficient secretion of the antidiuretic hormone (ADH) arginine vasopressin (AVP), is referred to as neurogenic (or central, neurohypophyseal, cranial, or hypothalamic) diabetes insipidus. Diabetes insipidus can also result from renal insensitivity to the antidiuretic effect of AVP, which is referred to as nephrogenic diabetes insipidus. Excessive water intake can result in polyuria, which is referred to as primary polydipsia; it can be due to an abnormality in the thirst mechanism, referred to as dipsogenic diabetes insipidus, or it can be associated with a severe emotional cognitive dysfunction, referred to as psychogenic polydipsia. Finally, the increased metabolism of vasopressin during pregnancy is referred to as gestational diabetes insipidus.
ARGININE VASOPRESSIN
Synthesis
The regulation of the release of AVP from the posterior pituitary is primarily dependent, under normal circumstances, on tonicity information relayed by central osmoreceptor neurons expressing transient receptor potential channel 1 (TRPV1) (Fig. 71.1)1 and peripheral osmoreceptor neurons expressing TRPV4.2 AVP and its corresponding carrier, neurophysin II, are synthesized as a composite precursor by the magnocellular neurons of the supraoptic and paraventricular nuclei of the hypothalamus.3 The precursor is packaged into neurosecretory granules and transported axonally in the stalk of the posterior pituitary. En route to the neurohypophysis, the precursor is processed into the active hormone. Preprovasopressin has 164 amino acids and is encoded by the 2.5 kb AVP gene located in chromosome region 20p13.4,5 The AVP gene (coding for AVP and neurophysin II) and the OXT gene (coding for oxytocin and neurophysin I) are located in the same chromosome region, at a very short distance from each other (12 kb in humans) in a head-tohead orientation. Data from transgenic mouse studies indicate that the intergenic region between the OXT and the AVP genes contains the critical enhancer sites for cell-specific expression in the magnocellular neurons.3 It is phylogenetically interesting to note that cis and trans components of this specific cellular expression have been conserved between the Fugu isotocin (the homolog of mammalian oxytocin) and rat oxytocin genes.6 Exon 1 of the AVP gene encodes the signal peptide, AVP, and the NH2-terminal region of neurophysin II. Exon 2 encodes the central region of neurophysin II, and exon 3 encodes the COOH-terminal region of neurophysin II and the glycopeptide. Provasopressin is generated by the removal of the signal peptide from preprovasopressin and from the addition of a carbohydrate chain to the glycopeptide (Fig. 71.2). Additional posttranslation processing occurs within neurosecretory vesicles during the transport of the precursor protein to axon terminals in the posterior pituitary, yielding AVP, neurophysin II, and the glycopeptide. The AVP-neurophysin II complex (Fig. 71.3) forms tetramers that can self-associate to form higher oligomers.7 Neurophysins should be seen as chaperonelike molecules facilitating intracellular transport in magnocellular cells. In the posterior pituitary, AVP is stored in vesicles. Exocytotic release is stimulated by minute increases in serum osmolality (hypernatremia, osmotic regulation) and by more pronounced decreases in extracellular fluid (hypovolemia, nonosmotic regulation). Oxytocin and neurophysin I are released from the posterior pituitary by the suckling response in lactating females.
Immunocytochemical and radioimmunologic studies have demonstrated that oxytocin and vasopressin are synthesized in separate populations of the supraoptic nuclei and the paraventricular nuclei neurons,8,9 the central and vascular projections of which have been described in great detail.10
Some cells express the AVP gene and other cells express the OXT gene. Immunohistochemical studies have revealed a second vasopressin neurosecretory pathway that transports high concentrations of the hormone to the anterior pituitary gland from parvocellular neurons to the hypophyseal portal system. In the portal system, the high concentration of AVP acts synergistically with corticotropin-releasing hormone (CRH) to stimulate adrenocorticotropic hormone (ACTH) release from the anterior pituitary. More than half of parvocellular neurons coexpress both CRH and AVP. In addition, while passing through the median eminence and the hypophyseal stalk, magnocellular axons can also release AVP into the long portal system. Furthermore, a number of neuroanatomic studies have shown the existence of short
portal vessels that allow communication between the posterior and anterior pituitary. Therefore, in addition to parvocellular vasopressin, magnocellular vasopressin is able to influence ACTH secretion.11,12
Some cells express the AVP gene and other cells express the OXT gene. Immunohistochemical studies have revealed a second vasopressin neurosecretory pathway that transports high concentrations of the hormone to the anterior pituitary gland from parvocellular neurons to the hypophyseal portal system. In the portal system, the high concentration of AVP acts synergistically with corticotropin-releasing hormone (CRH) to stimulate adrenocorticotropic hormone (ACTH) release from the anterior pituitary. More than half of parvocellular neurons coexpress both CRH and AVP. In addition, while passing through the median eminence and the hypophyseal stalk, magnocellular axons can also release AVP into the long portal system. Furthermore, a number of neuroanatomic studies have shown the existence of short
portal vessels that allow communication between the posterior and anterior pituitary. Therefore, in addition to parvocellular vasopressin, magnocellular vasopressin is able to influence ACTH secretion.11,12
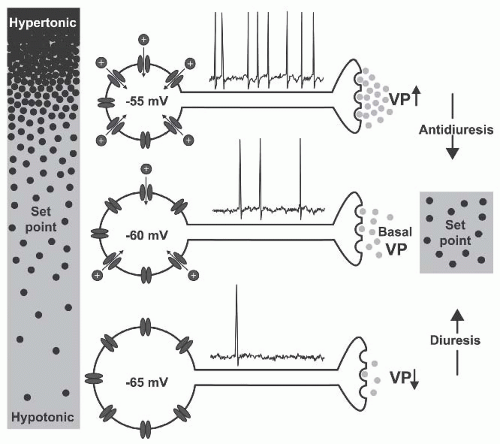 FIGURE 71.1 Osmoreception in vasopressin neurons. Changes in osmolality cause inversely proportional changes in soma volume. Shrinkage activates nonselective cation channels (NSCCs) and the ensuing depolarization increases the action potential firing rate and vasopressin (VP) release from axon terminals in the neurohypophysis. Increased VP levels in blood enhance water reabsorption by the kidney (antidiuresis) to restore extracellular fluid osmolality toward the set point. Hypotonic stimuli inhibit NSCCs.The resulting hyperpolarization and the inhibition of firing reduces VP release and promotes diuresis. (Modified from Prager-Khoutorsky and Bourque.15) |
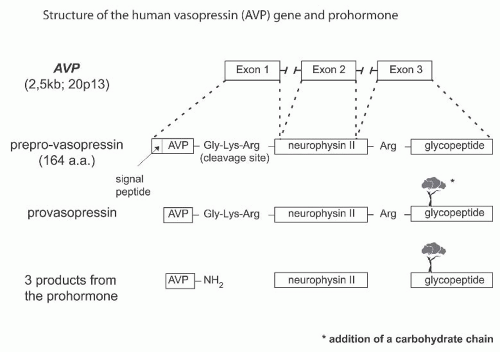 FIGURE 71.2 The structure of the human vasopressin (AVP) gene and prohormone. The cascade of vasopressin biosynthesis and signal peptide; AVP and arginine-vasopressin; neurophysin; and glycoprotein. |
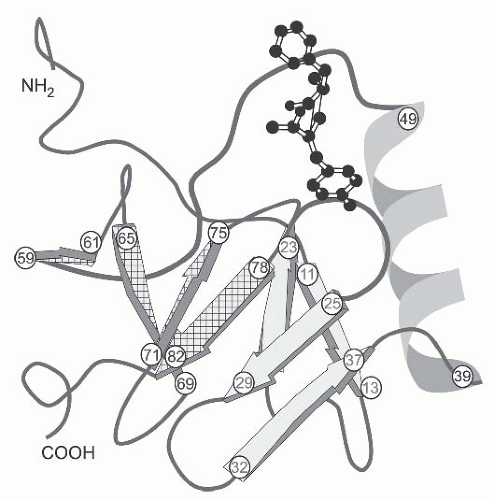 FIGURE 71.3 A three-dimensional structure of a bovine peptideneurophysin monomer complex.The structure of each chain is 12% helix and 40% β-sheet. The chain is folded into two domains as predicted by disulfide-pairing studies. The amino-terminal domain begins in a long loop (residues 1-10), then enters a fourstranded (residues 11-13, 19-23, 25-29, and 32-37) antiparallel β-sheet (sheet I; four solid arrows), followed by a three-turn 310-helix (residues 39-49) and another loop (residues 50-58). The carboxyl-terminal domain is shorter, consisting of only a fourstranded (residues 59-61, 65-69, 71-75, and 78-82) antiparallel β-sheet (sheet II; four cross-hatched arrows).7 The arginine vasopressin molecule (balls and sticks model) is shown in the peptide-binding pocket of the neurophysin monomer.The strongest interactions in this binding pocket are salt-bridge interactions between the αNH3+ group of the peptide, the γ-COO– group of GluNP47 (residue number 47 of the neurophysin molecule), and the side chain of ArgNP8. The γ-COO– group of GluNP47 plays a bifunctional role in the peptide-binding pocket: (1) it directly interacts with the hormone, and (2) it interacts with other neurophysin residues to establish the correct, local structure of the peptideneurophysin complex. ArgNP8 and GluNP47 are conserved in all neurophysin sequences from mammals to invertebrates. |
OSMOTIC AND NONOSMOTIC STIMULATION
The regulation of ADH release from the posterior pituitary is dependent primarily on two mechanisms involving the osmotic and nonosmotic pathways (Fig. 71.4).13 Although magnocellular neurons are themselves osmosensitive, they require input from the lamina terminalis to respond fully to osmotic challenges (Fig. 71.5). Neurons in the lamina terminalis are also osmosensitive and because the subfornical organ (SFO) and the organum vasculosum of the lamina terminalis (OVLT) lie outside the blood-brain barrier, they can integrate this information with endocrine signals borne of circulating hormones, such as angiotensin II (AT-II), relaxin, and atrial natriuretic peptide (ANP). Although circulating AT-II and relaxin excite both oxytocin and vasopressin magnocellular neurons, ANP inhibits vasopressin neurons.14
The nonosmotic pathways are more physiologically described now as “osmoregulatory gain.”15 A stable and approximately linear relation is normally observed between vasopressin concentration and plasma osmolality under resting conditions (Fig. 71.4A).16 The slope of this relation reflects the overall sensitivity of this homeostatic mechanism and the term “osmoregulatory gain” refers to this parameter. Osmoregulatory gain is increased during hypovolemia to help maintain arterial pressure and restore blood volume.17 Conversely, osmoregulatory gain is attenuated during hypervolemia to promote homeostasis by favoring diuresis (Fig. 71.6). The enhancement of osmosensory gain is mediated by circulating AT-II: neurons in the SFO (Fig. 71.5) contain AT-II and the release of this peptide during hypovolemia or hypotension can lead to an increase in the osmotic activation of magnocellular cells producing vasopressin. Good et al.18 and Zhang and Bourque19 found that AT-II enhances osmosensory gain by amplifying mechanosensory transduction, an F actin-dependent phenomenon probably part of a scaffolding complex.
In addition to an angiotensinergic path from the SFO, the OVLT and the median preoptic nucleus provide direct glutaminergic and GABAergic projections to the hypothalamoneurohypophyseal system. Nitric oxide may also modulate neurohormone release.3 The neuropeptide apelin is colocalized with AVP in supraoptic nucleus (SON) magnocellular neurons, and physiologic experiments indicate that AVP and apelin are conversely regulated to facilitate systemic AVP release and to suppress antidiuresis.20
The cellular basis for osmoreceptor potentials has been characterized using patch-clamp recordings and morphometric analysis in magnocellular cells isolated from the supraoptic nucleus of the adult rat. In these cells, stretchinactivating cationic channels transduce osmotically evoked changes in cell volume into functionally relevant changes in membrane potential. In addition, magnocellular neurons also operate as intrinsic Na+ detectors. The N-terminal variant of the TRPV1 is an osmotically activated channel expressed in the magnocellular cells producing vasopressin21 and in the circumventricular organs, the OVLT, and the SFO.22 Because osmoregulation still operates in Trpv1–/– mice, other osmosensitive neurons or pathways must compensate for the loss of central osmoreceptor function.21,22,23 Afferent neurons expressing the osmotically activated ion channel TRPV4 in the thoracic dorsal root ganglia that innervate hepatic blood
vessels and detect physiologic hypo-osmotic shifts in blood osmolality have recently been identified.2
vessels and detect physiologic hypo-osmotic shifts in blood osmolality have recently been identified.2
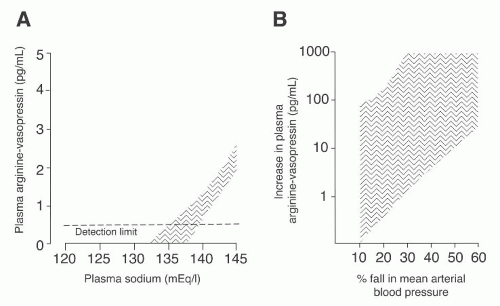 FIGURE 71.4 A: The osmotic and nonosmotic stimulation of arginine vasopressin (AVP).The relationship between plasma AVP (PAVP) and plasma sodium (PNa) in 19 normal subjects is described by the area with vertical lines, which includes the 99% confidence limits of the regression line PNa/PAVP.The osmotic threshold for AVP release is approximately 280 to 285 mmol per kilogram or 136 mEq of Na per liter. AVP secretion should be abolished when plasma sodium is less than 135 mEq per liter.214 B: The increase in plasma AVP during hypotension (vertical lines). Note that a large diminution in blood pressure in normal humans induces large increments in AVP. (From Zerbe RL, Henry DP, Robertson GL. Vasopressin response to orthostatic hypotension: etiological and clinical implications. Am J Med. 1983;74:265, with permission.) |
In mice lacking the osmotically activated ion channel TRPV4, hepatic sensory neurons no longer exhibit osmosensitive inward currents and the activation of peripheral osmoreceptors in vivo is abolished. In a large cohort of human liver transplantees, who presumably have denervated livers, plasma osmolality is significantly elevated compared to healthy controls, suggesting the presence of an inhibitory vasopressin effect of hyponatremia perceived in the portal vein from hepatic afferents.2 TRPV1 (expressed in central neurons) and TRPV4 (expressed in peripheral neurons) thus appear to play entirely complementary roles in osmoreception. McHugh et al.24 have therefore identified the primary afferent neurons that constitute the afferent arc of a well-characterized reflex in man, which was recently identified in rodents. This reflex engages the sympathetic nervous system to raise blood pressure and stimulate metabolism.25,26 Of clinical interest, it has already been demonstrated that orthostatic hypotension and postprandial hypotension respond to water drinking.27,28,29 Moreover, water drinking in man can prevent neutrally mediated syncope during blood donation or after prolonged standing.30 Finally, water drinking is also associated with weight loss in overweight individuals.31 Other peripheral sensory neurons expressing other mechanosensitive proteins may also be involved in osmosensitivity.32
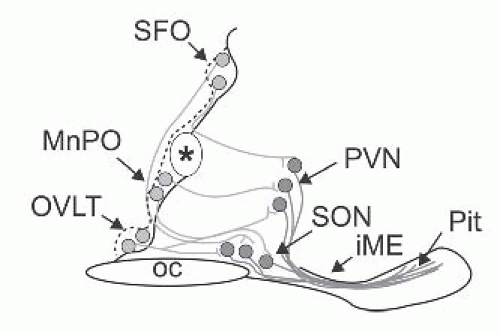 FIGURE 71.5 A schematic representation of the osmoregulatory pathway of the hypothalamus (sagittal section of midline of ventral brain around the third ventricle in mice). Neurons (lightly filled circles) in the lamina terminalis (OVLT), median preoptic nucleus (MnPO), and subfornical organ (SFO), which are responsive to plasma hypertonicity, send efferent axonal projections (grey lines) to magnocellular neurons of the paraventricular (PVN) and supraoptic nuclei (SON). The OVLT is one of the brain circumventricular organs and is a key osmosensing site in the mammalian brain (vide infra). The processes (darklines) of these magnocellular neurons form the hypothalamoneurohypophyseal pathway that courses in the median eminence to reach the posterior pituitary, where neurosecretion of vasopressin and oxytocin occurs. (Modified from Wilson et al., 2002 with permission, Copyright (2002), National Academy of Sciences USA.) |
The osmotic stimulation of AVP release by dehydration, hypertonic saline infusion, or both, is regularly used to determine the vasopressin secretory capacity of the posterior pituitary. This secretory capacity can be assessed directly by comparing the plasma AVP concentrations measured sequentially during the dehydration procedure with the normal values33 and then correlating the plasma AVP values with the urine osmolality measurements obtained simultaneously (Fig. 71.7).
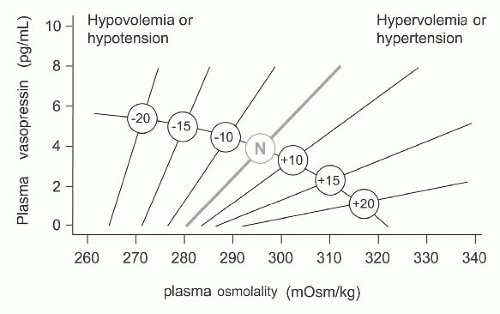 FIGURE 71.6 A schematic representation of the relationship between plasma vasopressin and plasma osmolality in the presence of differing states of blood volume and/or pressure. The line labeled N represents normovolemic normotensive conditions. Minus numbers to the left indicate a percent fall, and positive numbers to the right represent a percent rise in blood volume or pressure. Data from Vokes and Robertson.230 |
The AVP release can also be assessed indirectly by measuring plasma and urine osmolalities at regular intervals during the dehydration test.34 The maximal urine osmolality obtained during dehydration is compared with the maximal urine osmolality obtained after the administration of vasopressin (Pitressin, 5 U subcutaneously in adults, 1 U subcutaneously in children) or 1-desamino-8-D-arginine vasopressin (desmopressin [dDAVP], 1 to 4 µg s.c. or intravenously [IV], over 5 to 10 minutes).
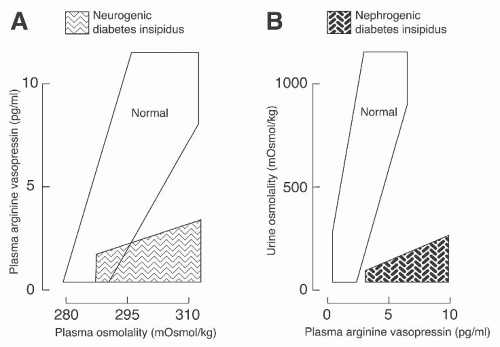 FIGURE 71.7 A: The relationship between plasma arginine vasopressin (AVP) and plasma osmolality during the infusion of a hypertonic saline solution. Patients with primary polydipsia and nephrogenic diabetes insipidus have values within the normal range (open area) in contrast to patients with neurogenic diabetes insipidus, who show subnormal plasma antidiuretic hormone (ADH) responses (cross-hatched area). B: The relationship between urine osmolality and plasma ADH during dehydration and water loading. Patients with neurogenic diabetes insipidus and primary polydipsia have values within the normal range (open area) in contrast to patients with nephrogenic diabetes insipidus, who have hypotonic urine despite high plasma ADH (stippled area). (Modified from Zerbe RL, Robertson GL. Disorders of ADH. Med North Am. 1984;13:1570.231) |
The nonosmotic stimulation of AVP release can be used to assess the vasopressin secretory capacity of the posterior pituitary in a rare group of patients with the essential hyponatremia and hypodipsia syndrome.35 Although some of these patients may have partial central diabetes insipidus, they respond normally to nonosmolar AVP release signals such as hypotension, emesis, and hypoglycemia.35 In all other cases of suspected central diabetes insipidus, these nonosmotic stimulation tests will not give additional clinical information.36
CLINICALLY IMPORTANT HORMONAL INFLUENCES ON THE SECRETION OF VASOPRESSIN
Angiotensin is a well-known dipsogen and has been shown to increase thirst in all the species tested.37 However, knockout models for angiotensinogen38 or for angiotensin-1A (AT-IA) receptor39,40 did not alter thirst or water balance. Disruption of the AT-II receptor induced only mild abnormalities of thirst postdehydration.41,42 However, as described earlier, AT-II enhances osmosensory gain. Earlier reports suggested that the intravenous administration of atrial peptides inhibits the release of vasopressin,43 but this was not confirmed by Goetz et al.44 Furthermore, Ogawa et al.45 found no evidence that ANP, administered centrally or peripherally, was important in the physiologic regulation of plasma AVP release in conscious rats. A very rapid and robust release of AVP is seen in humans after a cholecystokinin (CCK) injection.46 Nitric oxide is an inhibitory modulator of the hypothalamoneurohypophyseal system in response to osmotic stimuli.47,48,49,50 Vasopressin secretion is under the influence of a glucocorticoid-negative feedback system,51 and the vasopressin responses to a variety of stimuli (hemorrhage, hypoxia, hypertonic saline) in normal humans and animals appear to be attenuated or eliminated by pretreatment with glucocorticoids. Finally, nausea and emesis are potent stimuli of AVP release in humans and seem to involve dopaminergic neurotransmission.52
CELLULAR ACTIONS OF VASOPRESSIN
The neurohypophyseal hormone AVP has multiple actions, including the inhibition of diuresis, the contraction of smooth muscle, the aggregation of platelets, the stimulation of liver glycogenolysis, the modulation of adrenocorticotropic hormone release from the pituitary, and the central regulation of somatic functions (thermoregulation and blood pressure) and the modulation of social and reproductive behavior.53 These multiple actions of AVP can be explained by the interaction of AVP with at least three types of G protein-coupled receptors: the V1a (vascular, hepatic, and brain) and V1b (anterior pituitary) receptors act through phosphatidylinositol hydrolysis to mobilize calcium, and the V2 (kidney) receptor is coupled to adenylate cyclase.54,55,56
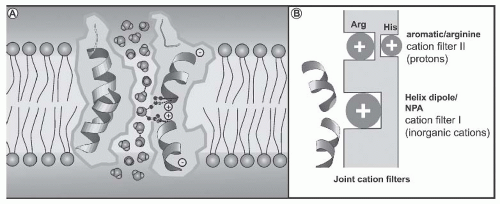 FIGURE 71.8 Schematic representations explaining the mechanism for blocking proton permeation of aquaporin 1 (AQP1). A: A diagram illustrating how partial charges from the helix dipoles restrict the orientation of the water molecules passing through the constriction of the pore. (From Murata K, Mitsuoka K, Hirai T, et al. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599.) B: A diagram illustrating how primordial AQPs selected against inorganic cations, such as Na+ and K+, because of a positive electrostatic field from the helix dipoles and the lack of cation coordination sites in the NPA region (filter I); yet, protons leaked through. Later, a second cation filter evolved in the ar/R region, which fully excluded protons (filter II) and provided individual selectivity properties for water, glycerol, urea, and ammonia.232 |
The transfer of water across the principal cells of the collecting ducts is now known at such a detailed level that billions of molecules of water traversing the membrane can be represented; see useful teaching tools at http://www.mpibpc.gwdg.de/abteilungen/073/gallery.html and http:// www.ks.uiuc.edu/research/aquaporins. The 2003 Nobel Prize in Chemistry was awarded to Peter Agre and Roderick MacKinnon, who solved two complementary problems presented by the cell membrane: How does a cell let one type of ion through the lipid membrane to the exclusion of other ions? And, how does it permeate water without ions? See Figure 71.8. This contributed to a momentum and renewed interest in basic discoveries related to the transport of water and indirectly to diabetes insipidus.57,58 The first step in the action of AVP on water excretion is its binding to arginine vasopressin type 2 receptors (hereafter referred to as V2 receptors) on the basolateral membrane of the collecting duct cells (Fig. 71.9). The human AVPR2 gene that codes for the V2 receptor is located in chromosome region Xq28 and has three exons and two small introns.59,60 The sequence of the cDNA predicts a polypeptide of 371 amino acids with seven transmembrane, four extracellular, and four cytoplasmic domains (Fig. 71.10). The activation of the V2 receptor on renal collecting tubules stimulates adenylyl cyclase via the stimulatory G protein (Gs) and
promotes the cyclic adenosine monophosphate (cAMP)-mediated incorporation of water channels into the luminal surface of these cells. There are two ubiquitously expressed intracellular cAMP receptors: (1) the classical protein kinase A (PKA) that is a cAMP-dependent protein kinase, and (2) the recently discovered exchange protein directly activated by cAMP that is a cAMP-regulated guanine nucleotide exchange factor. Both of these receptors contain an evolutionally conserved cAMPbinding domain that acts as a molecular switch for sensing intracellular cAMP levels to control diverse biologic functions.61 Several proteins participating in the control of cAMP-dependent aquaporin-2 AQP2 trafficking have been identifieD (e.g., A-kinase anchoring proteins tethering PKA to cellular compartments; phosphodiesterases regulating the local cAMP
level; cytoskeletal components such as F-actin and microtubules; small GTPases of the Rho family controlling cytoskeletal dynamics; motor proteins transporting AQP2-bearing vesicles to and from the plasma membrane for exocytic insertion and endocytic retrieval; soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors (SNAREs) inducing membrane fusions, hsc70, a chaperone important for endocytic retrieval). These processes are the molecular basis of the vasopressin-induced increase in the osmotic water permeability of the apical membrane of the collecting tubule.62,63,64
promotes the cyclic adenosine monophosphate (cAMP)-mediated incorporation of water channels into the luminal surface of these cells. There are two ubiquitously expressed intracellular cAMP receptors: (1) the classical protein kinase A (PKA) that is a cAMP-dependent protein kinase, and (2) the recently discovered exchange protein directly activated by cAMP that is a cAMP-regulated guanine nucleotide exchange factor. Both of these receptors contain an evolutionally conserved cAMPbinding domain that acts as a molecular switch for sensing intracellular cAMP levels to control diverse biologic functions.61 Several proteins participating in the control of cAMP-dependent aquaporin-2 AQP2 trafficking have been identifieD (e.g., A-kinase anchoring proteins tethering PKA to cellular compartments; phosphodiesterases regulating the local cAMP
level; cytoskeletal components such as F-actin and microtubules; small GTPases of the Rho family controlling cytoskeletal dynamics; motor proteins transporting AQP2-bearing vesicles to and from the plasma membrane for exocytic insertion and endocytic retrieval; soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors (SNAREs) inducing membrane fusions, hsc70, a chaperone important for endocytic retrieval). These processes are the molecular basis of the vasopressin-induced increase in the osmotic water permeability of the apical membrane of the collecting tubule.62,63,64
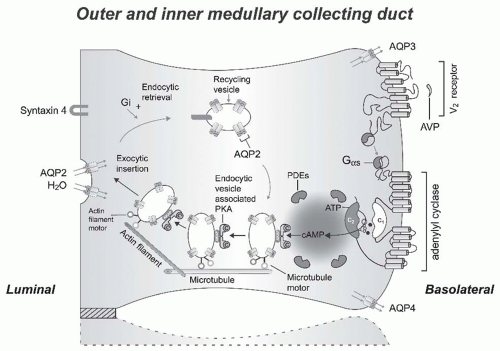 FIGURE 71.9 A schematic representation of the effect of vasopressin (AVP) to increase water permeability in the principal cells of the collecting duct. AVP is bound to the V2 receptor (a G-protein-linked receptor) on the basolateral membrane. The basic process of Gprotein-coupled receptor signaling consists of three steps: a heptahelical receptor that detects a ligand (in this case, AVP) in the extracellular milieu, a G-protein (Gαs) that dissociates into subunits bound to GTP and bg subunits after interaction with the ligand-bound receptor, and an effector (in this case, adenylyl cyclase) that interacts with dissociated G-protein subunits to generate small-molecule second messengers. AVP activates adenylyl cyclase, increasing the intracellular concentration of cAMP. The topology of adenylyl cyclase is characterized by two tandem repeats of six hydrophobic transmembrane domains separated by a large cytoplasmic loop and terminates in a large intracellular tail. The dimeric structure (C1 and C2) of the catalytic domains is represented. Conversion of ATP to cAMP takes place at the dimer interface. Two aspartate residues (in C1) coordinate two metal cofactors (Mg2+ or Mn2+ represented here as two small black circles), which enable the catalytic function of the enzyme.233 Adenosine is shown as an open circle and the three phosphate groups (ATP) are shown as smaller open circles. Protein kinase A (PKA) is the target of the generated cAMP. The binding of cAMP to the regulatory subunits of PKA induces a conformational change, causing these subunits to dissociate from the catalytic subunits. These activated subunits (C), as shown here, are anchored to an aquaporin-2 (AQP2)-containing endocytic vesicle via an A-kinase anchoring protein. The local concentration and distribution of the cAMP gradient is limited by phosphodiesterases (PDEs). Cytoplasmic vesicles carrying the water channels (represented as homotetrameric complexes) are fused to the luminal membrane in response to AVP, thereby increasing the water permeability of this membrane. The dissociation of the A-kinase anchoring protein from the endocytic vesicle is not represented. Microtubules and actin filaments are necessary for vesicle movement toward the membrane. When AVP is not available, AQP2 water channels are retrieved by an endocytic process, and water permeability returns to its original low rate. Aquaporin-3 (AQP3) and aquaporin-4 (AQP4) water channels are expressed constitutively at the basolateral membrane. |
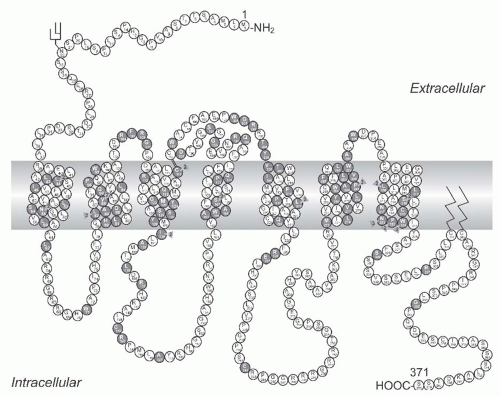 FIGURE 71.10 A schematic representation of the V2 receptor and identification of 193 putative disease-causing AVPR2 mutations. Predicted amino acids are shown as the one-letter amino acid code. A solid symbol indicates a codon with a missense or nonsense mutation; a number indicates more than one mutation in the same codon; other types of mutations are not indicated on the figure. There are 95 missense, 18 nonsense, 46 frameshift deletion or insertion, 7 in-frame deletion or insertion, 4 splice site, 22 large deletion mutations, and 1 complex mutation. |
AVP also increases the water reabsorptive capacity of the kidney by regulating the urea transporter UT-A1 that is present in the inner medullary collecting duct, predominantly in its terminal part.65,66 AVP also increases the permeability of principal collecting duct cells to sodium.67 Finally, the vasopressin V2 receptor has been located in the primary cilium and Bardet-Biedl syndrome-derived unciliated renal epithelial cells were unable to respond to luminal AVP and to activate luminal AQP2.68
In summary, in the absence of AVP stimulation, collecting duct epithelia exhibit very low permeabilities to sodium urea and water. These specialized permeability properties permit the excretion of large volumes of hypotonic urine formed during intervals of water diuresis. By contrast, AVP stimulation of the principal cells of the collecting ducts leads to selective increases in the permeability of the apical membrane to water, urea, and sodium.
These actions of vasopressin in the distal nephron are possibly modulated by prostaglandin E2 (PGE2), nitric oxide,69 and by luminal calcium concentration. High levels of E-prostanoid-3 receptors are expressed in the kidney.70 However, mice lacking E-prostanoid-3 receptors for PGE2 were found to have quasinormal regulation of urine volume and osmolality in response to various physiologic stimuli.70 PGE2 is synthesized and released in the collecting duct, which expresses all E-prostanoid receptors.71,72,73 An apical calcium/polycation receptor protein expressed in the terminal portion of the inner medullary collecting duct of the rat
has been shown to reduce AVP-elicited osmotic water permeability when luminal calcium concentration rises.74 This possible link between calcium and water metabolism may play a role in the pathogenesis of renal stone formation.74
has been shown to reduce AVP-elicited osmotic water permeability when luminal calcium concentration rises.74 This possible link between calcium and water metabolism may play a role in the pathogenesis of renal stone formation.74
KNOCKOUT MICE WITH URINARY CONCENTRATION DEFECTS
A useful strategy to establish the physiologic function of a protein is to determine the phenotype produced by pharmacologic inhibition of protein function or by gene disruption. Transgenic knockout mice deficient in AQP1, AQP2, AQP3, AQP4, AQP3, and AQP4; CLCNK1; NKCC2; AVPR2; AGT; or adenylyl cyclase 6 (AC6) have been engineered.75,76,77,78,79,80,81,82,83,84 Angiotensinogen (AGT)-deficient mice are characterized by both concentrating and diluting defects secondary to a defective renal papillary architecture.76
As reviewed by Rao and Verkman,85 the extrapolation of data in mice to humans must be made with caution. For example, the maximum osmolality of mice (greater than 3,000 mOsmol per kilogram of H2O) is much greater than that of human urine (1,000 mOsmol per kilogram of H2O), and normal serum osmolality in mice is 330 to 345 mOsmol per kilogram of H2O, substantially greater than that in humans (280 to 290 mOsmol per kilogram of H2O). These differences are related to renal anatomy and metabolic rate: (1) the mouse kidney exhibits much larger loops of Henle in the inner medulla and papillae and (2) because of the large difference in body size (30 g to 70 kg) the mouse metabolic rate (and thus the food intake) per gram of body mass is 20 times greater than that of humans. The amount of osmoles that need to be excreted by the kidney is also disproportionately larger when expressed per gram of body mass or kidney mass. These two features account for the much higher urine concentrating ability of the mouse as compared to humans. Protein expression patterns, and thus the interpretation of phenotype studies, may also be species dependent. For example, AQP4 is expressed in both the proximal tubule and the collecting duct in mice but only in the collecting duct in rats and humans.85
The Aqp3, Aqp4, Clcnk-1, and Agt knockout mice have no identified human counterparts. Of interest, AQP1-null individuals have no obvious symptoms.86 Yang et al.87 have generated an AQP2-T126M conditional knockin model of nephrogenic diabetes insipidus (NDI), to recapitulate the clinical features of the naturally occurring human AQP2 mutation T126M.88 The conditional knockin adult mice showed polyuria, urinary hypo-osmolality, and endoplasmic reticulum (ER) retention of AQP2-T126M in the collecting duct. The screening of candidate protein folding correctors in AQP2-T126M-transfected kidney cells showed increased AQP2-T126M plasma membrane expression with the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG), a compound currently in clinical trials for tumor therapy. 17-AAG increased urine osmolality in the AQP2-T126M mice (without effect in AQP2 null mice) and partially rescued defective AQP2-T126M cellular processing. These proof-of-concept findings suggest the possibility of using existing drugs for therapy of some forms of NDI. Ethylnitrosourea-mutagenized mice heterozygous for the F204V mutation in the Aqp2 gene have been described.89 The homozygous mice are viable because of a moderate phenotype with a possibility to concentrate urine from 161 to 470 m Osmol per kilogram of H2O in response to dDAVP. Cell biology experiments performed on renal tissue from Aqp2F204V/+ animals suggest that the mutant protein is being rescued by the wild-type protein.
Mice lacking the AVPR2 receptor failed to thrive and died within the first week after birth due to hypernatremic dehydration.78 Li et al.90 generated mice in which the Avpr2 gene could be conditionally deleted during adulthood by the administration of 4-OH-tamoxifen. Adult mice displayed all characteristic symptoms of X-linked NDI (XNDI), including polyuria, polydipsia, and resistance to the antidiuretic actions of vasopressin. Gene expression analysis suggested that the activation of renal EP4 PGE2 receptors might compensate for the lack of renal V2R activity in X-linked NDI mice, and both acute and chronic treatment of the mutant mice with a selective EP4 receptor agonist greatly reduced all major manifestations of XNDI. This beneficial effect is likely secondary to the intracellular generation of cAMP at the principal cell level by EP4 PGE2 receptors.
The absence of the gene coding for the NaK2Cl cotransport (NKCC2) in the luminal membrane of the thick ascending loop of Henle in the mouse also caused polyuria that was not compensated elsewhere in the nephron and recapitulated many features of the human classical Bartter syndrome.79 The absence of transcellular NaCl transport via NKCC2 probably abolished the lumen-positive transepithelial voltage that enables paracellular reabsorption of Na and K across the wall of the thick ascending tubule. The combined absence of transcellular and paracellular transport of salt across the thick ascending limb cells prevents the establishment of the normal osmotic gradient necessary for urine concentration.
EXPRESSION OF THE VASOPRESSIN GENE IN DIABETES INSIPIDUS RATS (BRATTLEBORO RATS)
The animal model of diabetes insipidus that has been most extensively studied is the Brattleboro rat. Discovered in 1961, the rat lacks vasopressin and its neurophysin, whereas the synthesis of the structurally related hormone oxytocin is not affected by the mutation.91 Its inability to synthesize vasopressin is inherited as an autosomal recessive trait. Schmale and Richter92 isolated and sequenced the vasopressin gene from homozygous Brattleboro rats, and found that the defect is due to a single nucleotide deletion of a G residue within the second exon encoding the carrier protein neurophysin (Fig. 71.11). The shift in the reading frame caused by this deletion predicts a precursor with an
entirely different C terminus. The messenger RNA (mRNA) produced by the mutated gene encodes a normal AVP but an abnormal NPII moiety,92 which impairs transport and processing of the AVP-NPII precursor and its retention in the endoplasmic reticulum of the magnocellular neurons where it is produced.93,94 Homozygous Brattleboro rats may still demonstrate some V2 (vide infra) antidiuretic effects since the administration of a selective nonpeptide V2 antagonist (SR 121463A, 10 mg per kilogram i.p.) induced a further increase in urine flow rate (200 to 354 ± 42 mL per 24 hours) and a decline in urinary osmolality (170 to 92 ± 8 mmol per kilogram).95 This decline in urine osmolality following the administration of a nonpeptide V2r antagonist could also be secondary to the “inverse agonist” properties of SR121463A: the intrinsic activity, or tone, of the V2R would be deactivated by the SR121463A compound (for the inverse agonist properties of SR121463A (see reference 96). There is also an alternative explanation to this relatively high urine osmolality of 170 because, in Brattleboro rats, low levels of hormonally active AVP are produced from alternate forms of AVP preprohormone. Due to a process called molecular misreading, one transcript contains a 2-bp deletion downstream from the single nucleotide deletion that restores the reading frame and produces a variant AVP preprohormone that is smaller in length by one amino acid and differs from the normal product by only 13 amino acids in the neurophysin II moiety.97 Oxytocin, which is present at enhanced plasma concentrations in Brattleboro rats, may be responsible for the antidiuretic activity observed.98,99 Oxytocin is not stimulated by increased plasma osmolality in humans.
entirely different C terminus. The messenger RNA (mRNA) produced by the mutated gene encodes a normal AVP but an abnormal NPII moiety,92 which impairs transport and processing of the AVP-NPII precursor and its retention in the endoplasmic reticulum of the magnocellular neurons where it is produced.93,94 Homozygous Brattleboro rats may still demonstrate some V2 (vide infra) antidiuretic effects since the administration of a selective nonpeptide V2 antagonist (SR 121463A, 10 mg per kilogram i.p.) induced a further increase in urine flow rate (200 to 354 ± 42 mL per 24 hours) and a decline in urinary osmolality (170 to 92 ± 8 mmol per kilogram).95 This decline in urine osmolality following the administration of a nonpeptide V2r antagonist could also be secondary to the “inverse agonist” properties of SR121463A: the intrinsic activity, or tone, of the V2R would be deactivated by the SR121463A compound (for the inverse agonist properties of SR121463A (see reference 96). There is also an alternative explanation to this relatively high urine osmolality of 170 because, in Brattleboro rats, low levels of hormonally active AVP are produced from alternate forms of AVP preprohormone. Due to a process called molecular misreading, one transcript contains a 2-bp deletion downstream from the single nucleotide deletion that restores the reading frame and produces a variant AVP preprohormone that is smaller in length by one amino acid and differs from the normal product by only 13 amino acids in the neurophysin II moiety.97 Oxytocin, which is present at enhanced plasma concentrations in Brattleboro rats, may be responsible for the antidiuretic activity observed.98,99 Oxytocin is not stimulated by increased plasma osmolality in humans.
 FIGURE 71.11 A neurophysin II genomic and amino acid sequence showing the 1 bp (G) deleted in the Brattleboro rat. The human sequence (GenBank entry M11166) is also shown. It is almost identical to the rat prepro sequence. In the Brattleboro rat, G1880 is deleted with a resultant frameshift after 63 amino acids (amino acid 1 is the first amino acid of neurophysin II). |
CLINICAL CHARACTERISTICS OF DIABETES INSIPIDUS DISORDERS
Central Diabetes Insipidus
Common Forms
Failure to synthesize or secrete vasopressin normally limits maximal urinary concentration and, depending on the severity of the disease, causes varying degrees of polyuria and polydipsia. Experimental destruction of the vasopressinsynthesizing areas of the hypothalamus (the supraoptic and paraventricular nuclei) causes a permanent form of the disease. Similar results are obtained by sectioning the hypophyseal-hypothalamic tract above the median eminence. Sections below the median eminence, however, produce only transient diabetes insipidus. Lesions to the hypothalamic-pituitary tract are often associated with a three-stage response both in experimental animals and in humans,100 which consists of:
1. An initial diuretic phase lasting from a few hours to 5 to 6 days.
2. A period of antidiuresis unresponsive to fluid administration. This antidiuresis is probably due to vasopressin release from injured axons and may last from a few hours to several days. Because urinary dilution is impaired during this phase, continued water administration can cause severe hyponatremia.
3. A final period of diabetes insipidus. The extent of the injury determines the completeness of the diabetes insipidus and, as already discussed, the site of the lesion determines whether the disease will or will not be permanent.
A detailed assessment of water balance following transsphenoidal surgery has been reported.101 There were 101 patients who underwent transsphenoidal pituitary surgery at the National Institutes of Health Clinical Center and were studied. Of the patients, 25% developed spontaneous isolated hyponatremia, 20% developed diabetes insipidus, and 46% remained normonatremic. Normonatremia, hyponatremia, and diabetes insipidus were associated with increasing degrees of surgical manipulation of the posterior lobe and pituitary stalk during surgery.
The etiologies of central diabetes insipidus in adults and in children are listed in Table 71.1.102,103,104 Rare causes of central diabetes insipidus include leukemia, thrombotic thrombocytopenic purpura, pituitary apoplexy, sarcoidosis, and Wegener granulomatosis.105 A distinctive syndrome characterized by early diabetes insipidus with subsequent progressive spastic cerebellar ataxia has also been described.106 Five patients who all presented with central diabetes insipidus and hypogonadism as first manifestations of neurosarcoidosis have been reported.107 Finally, circulating antibodies to vasopressin do not play a role in the development of diabetes insipidus.108 Antibodies to vasopressin occasionally develop during treatment with ADH and, when they do, almost always result in secondary resistance to its antidiuretic effect.108,109 Maghnie et al.102 studied 79 patients with central diabetes insipidus who were seen at four pediatric endocrinology units between 1970 and 1996. There were 37 male and 42 female patients whose median age at diagnosis was 7 years (range, 0.1 to 24.8 years). In 11 patients, central diabetes insipidus developed during an infectious illness or less than 2 months afterward (varicella in 5 patients,
mumps in 3 patients, and measles, toxoplasmosis, and hepatitis B in 1 patient each). Deficits in anterior pituitary hormones were documented in 48 patients (61%) a median of 0.6 year (range, 0.1 to 18.0 years) after the onset of diabetes insipidus. The most frequent abnormality was growth hormone deficiency (59%), followed by hypothyroidism (28%), hypogonadism (24%), and adrenal insufficiency (22%). Of the patients with histiocytosis of the Langerhans cells, 75% had an anterior pituitary hormone deficiency that was first detected at a median of 3.5 years after the onset of diabetes insipidus. The frequency and progression of histiocytosis of the Langerhans cells related to anterior pituitary and other nonendocrine hypothalamic dysfunction and their response to treatment in 12 adult patients has also been reviewed.110 None of the patients with central diabetes insipidus secondary to AVP mutations developed anterior pituitary hormone deficiencies.
mumps in 3 patients, and measles, toxoplasmosis, and hepatitis B in 1 patient each). Deficits in anterior pituitary hormones were documented in 48 patients (61%) a median of 0.6 year (range, 0.1 to 18.0 years) after the onset of diabetes insipidus. The most frequent abnormality was growth hormone deficiency (59%), followed by hypothyroidism (28%), hypogonadism (24%), and adrenal insufficiency (22%). Of the patients with histiocytosis of the Langerhans cells, 75% had an anterior pituitary hormone deficiency that was first detected at a median of 3.5 years after the onset of diabetes insipidus. The frequency and progression of histiocytosis of the Langerhans cells related to anterior pituitary and other nonendocrine hypothalamic dysfunction and their response to treatment in 12 adult patients has also been reviewed.110 None of the patients with central diabetes insipidus secondary to AVP mutations developed anterior pituitary hormone deficiencies.
| ||||||||||||||||||||||||||||||||||||||||||||||||||
Rare Forms
Inherited neurohypophyseal diabetes insipidus (OMIM 125700)111
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis




Related posts:
Computed Tomography and Magnetic Resonance Imaging
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Pathophysiology of Nephrotoxic Cell Injury
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
