Pathophysiology of Myeloma Cast Nephropathy
CN occurs mainly in patients with myeloma with a high LC secretion rate. That LCs are the main culprits is supported also by the following clinical, pathologic, and experimental data:
1. Renal lesions may recur on grafted kidneys.
2. Similar crystals may occasionally be seen within casts, proximal tubule cells, and plasma cells. Their usual lack of staining with anti-LC antibody is most likely due to degradation or masking of the relevant epitopes.
3. Mice injected with LC purified from patients with CN developed extensive cast formation in the distal renal tubules.
14
However, a number of patients produce large amounts of LCs and yet fail to present significant signs of renal involvement throughout the course of the disease. This may be related to the absence of enhancing factors (see later text), but this also suggests that some LCs may be particularly prone to induce renal lesions, especially cast formation.
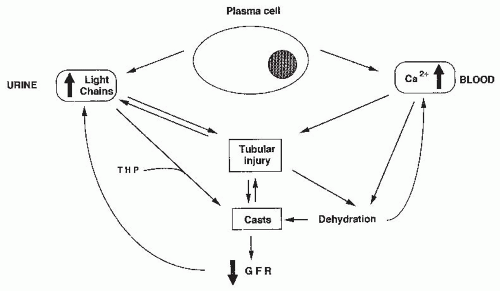
LCs are directly toxic to epithelial cells, resulting in decreased proximal reabsorption of the LCs and increased delivery to the distal tubule in which they coprecipitate with Tamm-Horsfall protein (THP). Tubular obstruction by large and numerous casts may also contribute to the development of tubular lesions. For clarity, we will analyze separately the pathogenesis of proximal tubule lesions that result from renal metabolism of LCs, the mechanisms of cast formation, and the respective role of tubular obstruction and tubular lesions in the genesis of renal failure (
Fig. 60.1).
Renal Metabolism of Light Chains and Pathogenesis of Proximal Tubule Lesions. Normal as well as malignant plasma cells can secrete free LC, in addition to complete Ig molecules; the amount of free LC secretion is highly variable, depending on the variable (
VL ) domain structure. The LCs are normally filtered by the glomerulus and then reabsorbed by the proximal tubule. Lambda and, to a lesser extent,
κ-LCs circulate mainly as covalently linked dimers that have a mass-restricted glomerular filtration. In normal individuals, several hundred milligrams per day of circulating free polyclonal LCs are filtered by glomeruli and more than 90% of these are reabsorbed and catabolized by proximal tubular cells. LCs bind to a single class of low affinity, high capacity noncooperative binding sites on both rat and human kidney brush-border membranes. These sites exhibit relative selectivity for LCs compared with albumin and β-lactoglobulin. It has been shown that LCs could bind to the tandem receptor cubilin
10 and megalin,
11 a multiligand receptor belonging to the large family of low density lipoprotein receptors, located in the intermicrovillar areas of the brush border. After binding to the luminal domain of proximal tubular epithelial cells, LCs are incorporated in endosomes that fuse with primary lysosomes where proteases, mainly cathepsin B, degrade the proteins into amino acids, which are returned to the circulation by the basolateral route.
When the concentration of filtered LCs is increased as in myeloma patients, profound functional and morphologic alterations of proximal tubule epithelial cells may occur. The functional disturbances include low molecular weight proteinuria and inhibition of sodium-dependent uptake of amino acids and glucose by brush-border preparations. Furthermore, in human proximal tubule cells, endocytosis of LCs was shown to induce activation of redox pathways,
31,
32 NF-κB,
33 and mitogen activated protein kinases (MAPK),
34 resulting in cytokine production including interleukin (IL)-6, IL-8, transforming growth factor (TGF)-
β, and monocyte chemoattractant protein-1 (MCP-1). Part of these effects may be mediated by megalin itself because megalin possesses intrinsic signaling properties. Moreover, excessive LC endocytosis may promote apoptosis
35 and induce epithelial-mesenchymal transition of tubular cells.
36,
37 Increased cytokine production may be a major mechanism mediating tubulointerstitial injury and progressive kidney disease in some patients with myeloma. Suppression of proinflammatory cytokine production using inhibition of p38 MAPK and translocation of NF-κB by pituitary adenylate cyclase- activating polypeptide with 38 residues (PACAP38) was shown to dramatically prevent injury of cultured renal proximal tubule cells caused by myeloma LCs.
38Morphologically, some of the LCs infused in mice or rats or perfused in rat nephrons in vivo accumulated in enlarged, distorded endosomes and lysosomes of the proximal convoluted tubule with frequent crystalloid formations. This was associated with mitochondrial alterations, focal loss of the microvillus border, and epithelial cell exfoliation.
Pathogenesis of Cast Formation. Because myeloma casts are composed principally of the monoclonal LC and THP/uromodulin, it has long been hypothesized that interaction of these two proteins was a key event in cast formation. THP is a highly glycosylated and acidic protein (isoelectric point [pI] = 3.2) synthesized exclusively by the cells of the ascending limb of the loop of Henle. It is the major protein constituent of normal urine, and an almost universal component of casts. This 80-kDa protein is also remarkable for its ability to form reversibly high-molecular-size aggregates of about 7 × 10
6 daltons at high but physiologic concentrations of sodium and calcium, and at low urinary pH. The role of THP in cast formation has prompted a wealth of studies on its interactions with LCs. These studies were performed with the aim of defining a population of myeloma patients at risk of developing renal damage. The role of LC pI has long been suggested. It was proposed that LC with a high pI (greater than 5.6) and THP could bear opposite charges in the normal urine pH range, and undergo polar interaction and precipitation. However, this hypothesis was not confirmed in further experimental and clinical studies.
25,
39
In a rat model, development of casts and injury to proximal tubule cells in renal tubules microperfused with human nephritogenic LC were not correlated with LC pI, molecular form, or isotype.
40 Intranephronal obstruction was aggravated by decreasing extracellular fluid volume or adding furosemide. In perfused loop segments, cast-forming LCs reduced chloride absorption directly, thereby increasing tubule fluid [Cl˜] and promoting their own aggregation with THP.
41 Pretreatment of rats with colchicine, which prevents addition of sialic acid to the protein, completely prevented obstruction and cast formation in perfused nephrons, and THP from those rats did not aggregate with LCs in vitro, contrary to THP purified from control rats. In vitro studies suggest that THP can undergo both self (homotypic) aggregation and heterotypic aggregation with LCs. Homotypic aggregation is enhanced by calcium, furosemide, and low pH, and is dependent on THP sialic-acid content. Heterotypic aggregation requires previous binding of LC to the THP protein backbone. A 9-residue sequence of the THP was identified as a binding site of LCs, including a histidine at position 226, which explains, at least partially, the pH dependence of molecular interactions.
42 LCs bind to THP through their third complementary determining region (CDR3).
43 The sugar moiety is also essential for coaggregation of LC and THP. THP from normal volunteers treated with colchicine had a lower sialic acid content and a decreased aggregation potential in the presence of pathogenic LCs. These findings suggest that colchicine may be useful in the treatment of cast nephropathy and that it is conceivable to design peptides or analogs that would inhibit interactions of LCs with THP and theoretically prevent myeloma CN.
Cast formation may not rely only on interactions between LC and THP. First, 5 of 12 LCs purified from the urine of patients with CN failed to react with THP.
16 Second, myeloma casts occasionally do not stain for THP in human
biopsies, and casts induced in mice by LC injection do not seem to contain THP during the first 24 hours, indicating that some LCs may undergo aggregation or precipitation in the absence of THP. This hypothesis is supported by studies showing that the deposition of certain LCs in vivo may be related to their capability to aggregate in vitro.
44 Resistance of LCs to renal and macrophage-released proteases may also contribute to cast formation and persistence.
16Role of Tubular Obstruction by Casts in the Genesis of Renal Failure. The role of casts as plugs obstructing the tubules has been clearly shown in micropuncture studies. In myeloma patients, the correlation between severity of renal insufficiency and the number of casts remains controversial.
24,
25,
45 This may be explained partly by the prominent medullary localization of casts, the count of which is underestimated in superficial kidney cortex biopsy specimens. The first indication that antibodies to THP could serve as probes of tubular obstruction was provided by Cohen and Border,
46 who identified the protein in glomerular urinary spaces of two myeloma patients. This finding is indicative of intratubular urinary backflow. We detected THP in glomerular urinary spaces in 16 of 18 biopsies of patients with myeloma CN (Ronco and Mougenot, personal data) (
Fig. 60.2). The proportion of obstructed tubules is too small to account by itself for renal failure. Renal failure induced by CN is multifactorial, implicating also tubular epithelial cell and interstitial lesions. Tubule obstruction by casts may explain the slow recovery of renal function noted in many patients.
25
Interstitial deposits of THP were also found in 8 (44%) of the 18 biopsies (Ronco and Mougenot, personal data). They probably result from a leakage of the protein through gaps in the tubular basement membrane favored by tubular obstruction. Clinical and experimental models have implicated the protein in the pathogenesis of tubulointerstitial nephritis. Thomas et al.
47 identified a single class of sialic acid-specific cell surface receptors for THP on polymorphonuclear leukocytes, and further showed that in vitro activation of human mononuclear phagocytes by particulate THP led to the release of gelatinase and reactive oxygen metabolites, both probably contributing to tissue damage.
Clinical Presentation
Changing Presentation of Patients with Myeloma-Induced Renal Failure. When DeFronzo et al. reported the first series of 14 myeloma patients with acute renal failure in 1960,
48 it was established that renal failure occurred at some time during the illness in approximately half of the patients, but that the mode of presentation was usually chronic with a slow progression over a period of several months to years.
The mode of presentation of renal failure in myeloma has changed dramatically over the years. In their review of 141 patients treated in Nottingham between 1960 and 1988, Rayner et al.
49 showed that the absence of severe renal impairment at presentation predicted a low probability of developing renal failure subsequently. In only 5 of 34 patients of our own renal series
25 did the diagnosis of myeloma antedate the discovery of renal failure by more than 1 month. In three patients, the presence of a monoclonal Ig was known for 10 to 18 years, but it only showed criteria of malignancy for less than 9 months. Two-thirds of the 107 patients referred to the Oxford Kidney Unit from 1987 to 2006
50 had myeloma diagnosed after their admission with acute kidney injury (AKI). More aggressive treatment of myeloma and higher awareness of the conditions that induce CN in the last two decades may have prevented LC precipitation within the tubule lumen in the patients with an established diagnosis of myeloma.
Demographic and Hematologic Characteristics of Patients with Cast Nephropathy-Related Renal Failure. Table 60.2 summarizes the clinical and pathologic data in four large series of myeloma patients with acute renal failure in which a renal biopsy was performed in at least 40% of the patients. A diagnosis of myeloma CN was established histologically in 81 of 99 (82%) renal biopsies, and lesions compatible with this diagnosis were found in 10 further biopsy specimens (10%). In comparison with the Mayo Clinic series of 869 unselected myeloma cases
52 in which the mean age was 62 years and the male-female ratio was 1.55, patients with acute renal failure did not show any demographic particularity. Myeloma patients with renal failure are characterized by high tumor mass and virtually constant urinary LC loss, often of high output.
More than 70% of patients in the renal series have a high tumor burden (
Table 60.2). This is confirmed by the
Alexanian series, which included 494 consecutive patients referred to an oncology center (
Table 60.3).
53 Only 3% of patients with myeloma of low tumor mass had renal failure, whereas 40% of those with high tumor burden had a serum creatinine greater than 180 µmol/L. These data contrast with the hematologic characteristics of patients with other renal complications of dysproteinemia including FS, amyloidosis, and MIDD, in whom the monoclonal B lymphocyte or plasma cell proliferation is either malignant but usually of low magnitude, or often benign from a hematologic point of view.
Another salient feature of myeloma associated with renal failure is the high prevalence of pure LC myelomas. Although they represent only about 20% of all myelomas, they are found in between 37% and 64% of patients with renal failure of presumed or established tubulointerstitial origin. Development of CN in two studies in which this diagnosis was established histologically
25,
27 was associated with urinary excretion of LCs exceeding 2 g per day in 53% and 72% of the patients (
Table 60.2). LC protein excretion emerges as a highly significant independent factor of renal failure on multivariate analysis (
Table 60.4). The risk of developing renal failure is twice as high in patients with pure LC myeloma, and five to six times greater in patients with LC proteinuria greater than 2.0 g per day compared to those with proteinuria less than 0.05 g per day. This indicates that in patients producing complete Ig molecules, CN essentially occurs in those synthetizing an excess of LCs. The frequency of renal failure is identical in patients excreting
κ or
λ λ LCs. IgD myeloma has the greatest potential for causing renal disease.
50 Hypercalcemia also is a prominent independent pathogenetic factor on multivariate analysis, with a risk of renal failure five times greater in those patients with corrected calcium greater than 2.87 mmol/L.
53The Clinical and Urinary Syndrome of Myeloma Cast Nephropathy. CN-induced renal failure is remarkably silent. Clinical signs are due to myeloma (or to hypercalcemia), including weakness, weight loss, bone pain, and infection. Because of their nonspecificity and their frequency in older patients, they often do not lead patients to take medical advice or physicians to prescribe serum and urinary electrophoreses, which are the key laboratory investigations for the diagnosis of myeloma. Peaks visible on serum or urine electrophoresis are then identified by immunoelectrophoresis or immunofixation. A preserved corrected calcium at presentation in patients with unexplained renal failure should alert clinicians to the possibility of myeloma.
The main urinary feature is the excretion of a monoclonal LC, which accounts for 70% or more of total proteinuria in 80% of patients.
25 LC proteinuria is usually not detected by urinary dipsticks, but only by techniques measuring total proteinuria. Certain LCs fail to react or react weakly in some widely used precipitation assays, such as the sulfosalicylic acid method, leading to falsely negative or underestimated results. The remaining proteins are composed of albumin and low molecular weight globulins that have failed to be reabsorbed by proximal tubule cells. In the rare patients with albuminuria greater than 1 g per day, CN is usually associated with glomerular lesions due to amyloidosis or MIDD. There is no hematuria in pure CN.
Precipitants of Cast Nephropathy. These are of paramount importance because of measures to prevent precipitation (
Table 60.5). It is often difficult to identify a particular event responsible for precipitating renal failure, as these patients experience many of the complications of the disease at once, a common thread of which seems to be an effect on renal perfusion.
Hypercalcemia is an important precipitant found in 16% to 44% of the renal series (
Table 60.5), and in 57% of the patients with renal failure in Alexanian nonrenal series.
53 Presumably, hypercalcemia acts by inducing dehydration as a result of emesis and a nephrogenic diabetes insipidus. It may also enhance LC toxicity and cause nephrocalcinosis.
Dehydration, with or without hypercalcemia, and infection are other major risk factors for acute renal failure. Rota et al.
25 found a high rate of urinary infections (10/34, 29%), which were associated in three cases with an increased proportion of polymorphonuclear leukocytes in the renal biopsy, suggesting an etiologic link between infection and deterioration of renal function. Infection also operates by causing dehydration and prompting the use of nephrotoxic antibiotics.
Contrast media have hitherto been considered an important precipitant of acute renal failure. It was hypothesized that the contrast medium bound to intratubular proteins, especially the LC and THP, causing them to precipitate and obstruct tubular flow. Contrast media also have vasoconstrictive effects, decreasing GFR and urinary output. McCarthy and Becker
54 reviewed seven retrospective studies of myeloma patients receiving contrast media, involving 476 patients who had undergone a total of 568 examinations. The prevalence of acute renal failure (which was not defined) was 0.6% to 1.25%, compared to 0.15% in the general population. This is a low risk and contradicts the dogma that contrast media should not be used in myeloma patients. This change may reflect awareness of the risk and care taken to hydrate patients actively with alkaline solutes before and during the administration of contrast media. No clinical data currently support the preferential use of nonionic agents in myeloma patients to decrease the risk of acute renal failure.
A number of drugs are noxious in myeloma patients. They include antibiotics, particularly aminoglycosides, and nonsteroidal anti-inflammatory drugs (NSAIDs).
25,
50 NSAIDs reduce the production of vasodilatory prostaglandins that help to maintain an appropriate GFR in patients with renal hemodynamics compromised by dehydration. Angiotensinconverting enzyme (ACE) inhibitors can also precipitate renal failure because they reduce GFR dramatically in dehydrated patients. Their use as that of angiotensin type-1 receptor antagonists should be avoided as long as a risk of decreased renal perfusion persists.
Recently introduced therapies including bisphosphonates may also induce toxic tubular injury. Renal failure secondary to acute tubular necrosis was reported with zoledronate, a potent bisphosphonate that is in widespread use for the treatment of hypercalcemia of malignancy,
55 and with short-term, high-dose pamidronate.
56 Doses should be adapted to GFR to avoid toxicity.
Renal Pathology and the Value of Kidney Biopsy
A kidney biopsy should not be routinely performed in patients with a presumed diagnosis of myeloma CN. However, it is useful in three circumstances:
1. To establish the cause of renal failure in anuric patients with clinically silent myeloma without evidence of serum monoclonal component on electrophoresis;
2. To analyze tubulointerstitial lesions and predict the reversibility of renal failure in patients with presumed CN but multiple precipitating factors;
3. To identify glomerular lesions in patients with urinary albumin greater than 1 g per day and no evidence of amyloid deposits in “peripheral” biopsies (accessory salivary glands, rectum, abdominal fat).
A kidney biopsy should be systematically performed in patients enrolled in therapeutic protocols because renal lesions should be precisely identified.
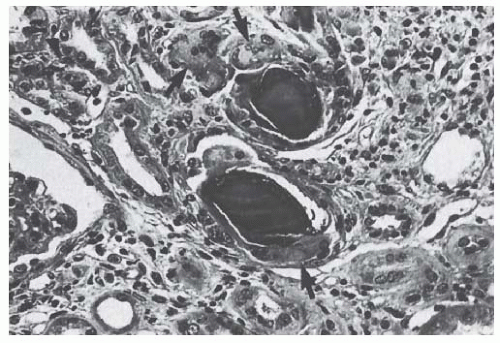
57Myeloma Casts. Myeloma CN is characterized by the presence of specific casts associated with severe alterations of the tubule epithelium. Myeloma casts are large and usually numerous. Their prevailing localization is the distal tubule and the collecting duct, but they may also be found in the proximal tubule and even in the glomerular urinary space. They often have a “hard” and “fractured” appearance, and show polychromatism upon staining with Masson’s trichrome (
Fig. 60.3). Casts may also have a stratified or laminated appearance. They may stain with Congo red, but only exceptionally do they show the typical yellow-green dichroism of amyloid under polarized light.
An important diagnostic feature of myeloma casts is the presence of crystals, which may be suspected by light microscopy.
58 Such casts are often angular or heterogeneous because they contain multiple rhomboid or needle-shaped crystals surrounded by amorphous material and cell debris.
Casts are frequently surrounded by mononuclear cells, exfoliated tubular cells, and, more characteristically, by multinucleated giant cells whose macrophagic origin has been established by specific antibodies. These cells are often seen engulfing the casts and at times actually phagocytizing fragments. In some cases, the cellular reaction is made
of polymorphonuclear leukocytes in the absence of urinary tract infection. Typical myeloma casts with a giant, multinucleated cell reaction (
Fig. 60.3) can be very occasionally detected in other hemopathies including
µ-HC disease
30 and Waldenstöm’s macroglobulinemia.
29 In myeloma CN, there is a great variability in the respective percentage of typical myeloma casts and of nonspecific hyaline casts. In some instances, most casts have nonspecific characteristics by light microscopy, even if by immunofluorescence the vast majority consists predominantly of one of the two LC types. The search for typical casts has to be conducted on all available sections if necessary.
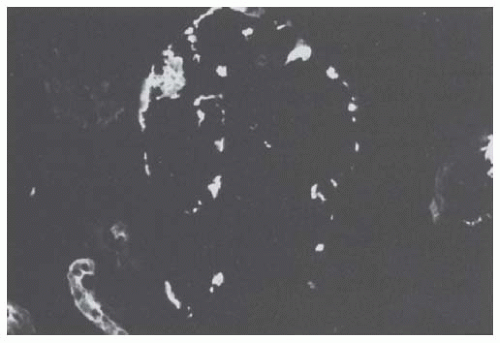
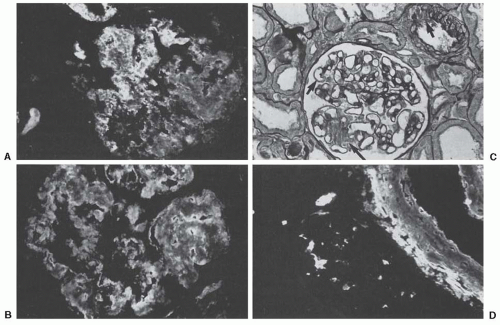
By immunofluorescence, myeloma casts are essentially composed of the monoclonal LC excreted by the patient, together with THP. In most cases, casts are stained exclusively or predominantly with either the anti-
κ or the anti-κ antibody. However, in about 25% of myeloma biopsies, casts stain for both antibodies because they contain polyclonal LCs, together with albumin and fibrinogen.
58 Staining of “angular” casts is often irregular, and more intense at the periphery (
Fig. 60.4). In heterogeneous casts, the crystals themselves fail to stain, whereas the matrix of the cast and the surrounding cellular debris and amorphous material often stain positively for one of the LC isotypes.
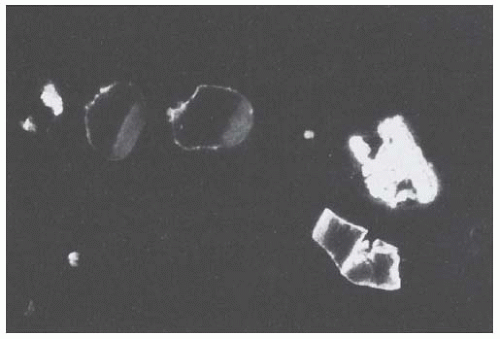
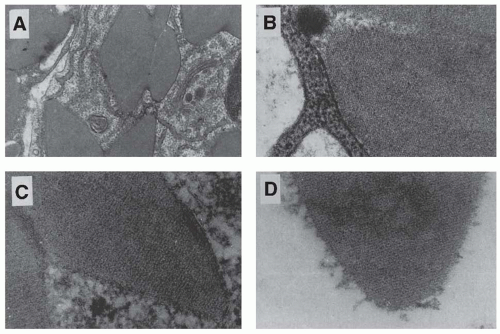
Cast ultrastructure was studied by electron microscopy in 24 biopsies of myeloma CN by Pirani et al.
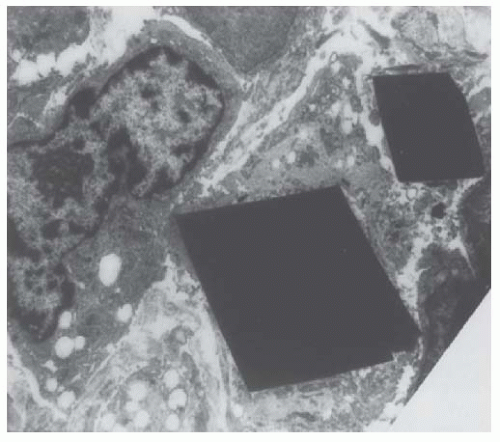
58 Crystals were detected in 14 biopsy specimens and suspected in another 4. The authors have identified four major categories of casts, according to their content and ultrastructural appearance. One category characterized by large rectangular crystals, or fragments thereof, with a pentagonal or hexagonal cross-section, is found only in myeloma CN. It seems to be closely linked to the development of a giant cell reaction around the cast. A second category also frequently contains crystals, but they are small, electron-dense, and needle-shaped, and seemingly not associated with a cellular reaction. Similar large rectangular and small, needle-shaped crystals can be found within plasma cells. They are also seen occasionally within the cytoplasm of either proximal or distal tubular cells (
Fig. 60.5), surrounded by a single smooth membrane, which suggests that they are located within lysosomes.
Tubules and Interstitium. Considerable tubular damage is almost always present in myeloma CN. Epithelial tubular lesions are not only seen in the distal tubules where casts are principally located, but also in proximal convoluted tubules, where the epithelium undergoes atrophy and degenerative changes. Frank tubular necrosis may also be seen, with or without typical myeloma CN.
25 By immunofluorescence, a variable number of tubule sections contain numerous “protein reabsorption droplets” staining for the monoclonal LC.
46
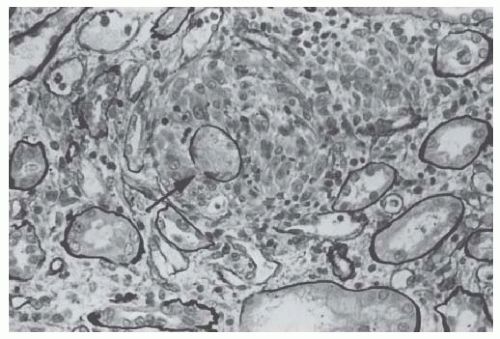
Interstitial lesions are often associated with the tubular damage. They may be mild and consist of inflammatory infiltrates and fibroedema, but fibrosis and its correlate, tubular atrophy, may also be fairly extensive. In severe cases with epithelial denudation and gaps in the continuity of the tubular basement membrane, often in close contact with myeloma casts, granulomatouslike formations containing macrophages and histiocytes develop around the ruptured tubules (
Fig. 60.6).
46Glomeruli and Vessels. The glomeruli are usually normal, except for small clusters of globally sclerotic glomeruli and a mild thickening of the mesangial matrix. When mesangial thickening is more prominent, the possibility of an associated
MIDD should be considered. Rarely, amorphous deposits reminiscent of myeloma casts can be seen in capillary loops or in the glomerular urinary space. In younger patients, severe chronic vascular lesions are sometimes observed, which may contribute to progression of sclerosis.
Outcome and Prognosis of Myeloma Cast Nephropathy
Until the 1980s, myeloma-induced renal failure was associated with a very poor prognosis, with a median survival of less than 1 year.
48 In recent years, the outcome of patients with myeloma, including those with renal impairment, has improved with the introduction of novel therapies, including high dose therapy followed by autologous stem cell support, and development of new drugs with a strong anti-myeloma effect (bortezomib, thalidomide, lenalidomide).
59 However, because patients with elevated serum creatinine levels were excluded from most randomized controlled studies, optimal treatment of multiple myeloma with renal failure remains to be defined. The establishment of consensus criteria for the assessment of renal function and renal response in multiple myeloma, and the use of modern sensitive tests to evaluate hematologic response, such as as nephelometric assays for serum free LC,
60 should help to improve renal and patient outcomes in the future.
61Renal Outcome and Prognostic Factors. Renal prognosis in patients with myeloma who present with renal failure remains poor, as complete or partial renal recovery occurs in half of patients after weeks to months,
62 and in only 20% to 40% of those with dialysis-dependent renal failure.
63,
64,
65,
66 Elevated plasma creatinine concentration or decreased estimated GFR (calculated using the MDRD equation) have been quoted as markers of poor renal prognosis in most studies,
26,
28,
62,
65,
67 implying that renal functional impairment of any degree should be treated as a medical emergency. In the study by Rota et al.,
25 main prognostic indicators were provided by renal histology. Renal response was seen in patients with typical cast nephropathy and/or tubular necrosis without interstitial damage. Global tubular atrophy and interstitial fibrosis were associated with partially or totally irreversible renal failure, whereas the number of casts has a controversial predictive value.
25,
26,
68
The rapid achievement of sustained hematologic response appears as a key factor for renal prognosis.
57,
65,
69,
70 In three recent studies a minimum of 50% reduction in serum free LC concentration was required for recovery of renal function in patients with biopsy proven CN.
57,
70,
71 Recent data indicate that outcome of severe renal failure may be substantially improved by bortezomib plus dexamethasone-based chemotherapy
61,
65,
66,
72 and, in patients requiring dialysis, by extended hemodialysis using a high-cut off dialyzer that allows effective removal of LCs.
70,
73Survival and Predictors. Myeloma patients with renal failure have a shorter survival than those with normal renal function. In the presence of renal failure, mortality in the first 3 months is about 30%,
26,
28,
67 and median survival ranges from 9 to 22 months. However, several studies have indicated that recovery of renal function is associated with improved survival, close to that of patients who do not develop renal failure.
25,
26,
27,
28,
50,
67,
62,
69 A response to chemotherapy is a key predictor of renal outcome and patient survival.
Fanconi Syndrome
Fanconi syndrome (FS) is characterized by renal glycosuria, generalized aminoaciduria, hypophosphatemia, and, frequently, by chronic acidosis, hypouricemia, and hypokalemia. It often includes osteomalacia, with pseudofractures. These manifestations result from functional impairment of the renal proximal tubule. The first association of FS with myeloma was reported by Sirota and Hamerman,
99 although these authors considered FS and myeloma as two separate diseases. Engle and Wallis
100 identified crystal-like inclusions in both tumor cells and renal tubule epithelial cells, and suggested that FS and myeloma could be related. Costanza and Smoller
101 described the cytoplasmic inclusions as round or rodlike electron-opaque structures with longitudinally oriented fibrils. Lee et al.
102 established clearly that mye loma was a cause of adult FS. Maldonado et al.
103 reported 17 cases of FS associated with plasma cell dyscrasia, and two more recent studies described the clinicopathologic features of the disease in two series of 11 and 32 patients.
104,
105 The disease is most likely underdiagnosed. The rarity of FS in patients with myeloma contrasts, however, with the high prevalence of tubule alterations in myeloma autopsy series. This suggests that unusual specific properties of LCs, mostly
κ, are involved in the pathophysiology of FS.
Pathophysiology of Plasma Cell Dyscrasia-Associated Fanconi Syndrome
The peculiar propensity of certain LCs to form crystals in vivo is attested by experimental studies in mice
14 and rats.
40 It is remarkable that the
κLCs that induced crystallization in vivo, also significantly reduced the glucose, chloride, and volume fluxes.
Crystal composition was analyzed in a patient with myeloma-associated FS and hexagonal crystals in kidney proximal tubular cells, bone marrow plasma cells, and phagocytes.
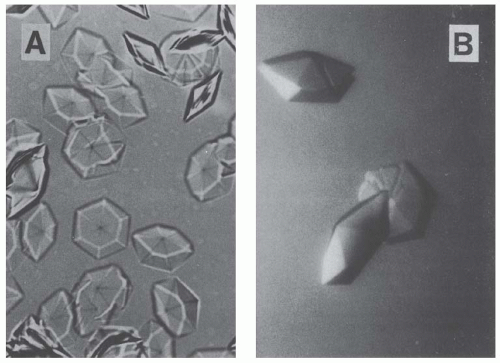
106 N-terminal sequencing and mass spectrometry studies showed that a 107-amino acid fragment corresponding to the variable domain of the κ-LC (V#c) was the essential component of crystals forming spontaneously from the patient’s urine (
Fig. 60.7A). Vκ was also crystallized alone using the hanging drop technique (
Fig. 60.7B). Crystals were hexagonal bipyramids and had the same 6.0-nm periodicity on electron micrographs as those found in the cells. The V domain (12-kDa) resisted proteolysis by trypsin, pepsin, and cathepsin B, self-reacted, and formed crystals in vitro, which may explain its accumulation in plasma cells and proximal tubular cells. The resistance of LC V domains to proteolytic enzymes including cathepsin B was confirmed in further studies,
16,
104 except in a few patients with a high-mass myeloma or Waldenström’s macroglobulinaemia
107 and FS. At variance with the observations made in patients with CN,
16 LCs from patients with FS did not bind THP, except in one case where both syndromes were associated.
The unusual physicochemical behavior of FS κ-chains was tentatively correlated with their structure in a number of cases.
108 Sequence analyses showed that 90% of LCs belonged to the VλVI variability subgroup, whereas this subgroup only accounts for 56% of all monoclonal κ-LCs.
104,
109 The VλVI appeared to originate from only two germline genes, IGKV1-39 in five cases and IGK1-33 in four. Analyses of the DNA sequence suggested that all structure peculiarities arose from somatic mutations in the proliferating clone.
110 In the 10 available sequences, residues had never or rarely been reported among VλVI subgroup LCs. The unusual presence of nonpolar or hydrophobic amino acids in the complementary determining region (CDR)-L1
loop at position 30, together with a nonpolar amino acid at position 50, seems to be specific for FS LCs derived from gene IGKV1-39. These hydrophobic residues are exposed to the LC surface
108 and may be involved in the pathophysiology of FS, as is suggested by site-directed mutagenesis in an experimental model for FS.
111 Recently, a transgenic mouse model with overexpression of human FS κ LC (CHEB) was generated through insertion of the V domain from CHEB LC in the Igκ locus of the mouse. This resulted in the expression of a hybrid κ LC made up of the human V domain and the mouse constant (C) region. Despite the replacement of the human C region, animals exhibited characteristic LC crystals within proximal tubular cells. This model confirmed that LC aggregation in FS is promoted by the V domain structure. The extent of tubular inclusions was proportional to the production rate and serum levels of CHEB LC. Using an inducible CRE mediated deletion of the CHEB V domain, tubular lesions were shown to recover after a few weeks when the LC production was stopped.
112After endocytosis, LCs are processed in the endosomal and lysosomal compartment where “normal” LCs are degraded. In FS, accumulation of the protease-resistant V domain fragment generated by lysosomal enzymes may induce crystal formation. Clogging of the endolysosomal system may subsequently alter apical membrane recycling and/or adenosine triphosphate (ATP) production (hence, Na
+-κ
+-ATPase functioning) as suggested by mitochondrial injury,
102 and lead to progressive impairment of sodium-dependent apical transporters. However, in a few cases of FS, crystalline inclusions were not observed within proximal tubular cells.
104,
107,
108 In two patients, LCs, which belonged to the VkIII subgroup, showed no common substitution with previously described FS VκI LC and did not display resistance to proteolysis, suggesting that other mechanisms of toxicity may be involved in the pathogenesis of the disease.
10 Furthermore, why FS does not occur in patients with apparently the same degree of distortion of the lysosomal compartment as can be seen in certain myeloma patients with or without CN is unclear. The molecular mechanisms responsible for glycosuria, phosphaturia, generalized aminoaciduria, and uric acid loss remain poorly understood. An impairment of the megalin-cubilin system might be involved.
Clinical Presentation
The clinical features are summarized in
Table 60.6.
104 The median age at diagnosis is 57 years. Most common initial manifestations are bone pain and weakness, principally due to osteomalacia. The major cause of this osteomalacia is hypophosphatemia, which results from increased urinary clearance of phosphate. Chronic acidosis and abnormal renal vitamin D metabolism further contribute to the development of bone lesions. Bone pain may also be the consequence of lytic lesions in patients with a high-mass myeloma. Other revealing signs are essentially due to the proximal tubule impairment, including hypokalemia. Renal failure occurs more frequently than one would expect in a disease of the proximal tubule.
Criteria for the diagnosis of FS may not all be present together, especially in patients with renal failure.
109 The diagnosis of FS is often unrecognized for several years in patients presenting with proteinuria, bone pain, or renal failure. The mean time from onset to diagnosis of FS is about 3 years.
104 Typically, the diagnosis of FS precedes that of the plasma cell dyscrasia, most often a κ-LC-excreting multiple
myeloma, because the hematologic disease has a low tumor burden and a slow progression. In 35 of 98 (36%) published cases,
104,
105 even criteria for the diagnosis of myeloma were lacking, and patients were classified initially as having a benign monoclonal gammopathy of undetermined significance (MGUS). In some patients, the diagnosis of the plasma cell dyscrasia remained undetermined between myeloma and MGUS because it may be difficult to recognize the cytologic characteristics of myeloma cells when their cytoplasm is stuffed with crystals. Three patients of 99 had Waldenström’s macroglobulinemia.
104,
105,
107Conversely, the metabolic disorders of FS may be overseen in the context of myeloma, and FS-related bone lesions should not be interpreted as the consequence of high-mass myeloma.
Pathologic Data
Typically there are prominent crystals in enlarged proximal tubular cells and degenerative changes of proximal tubules.
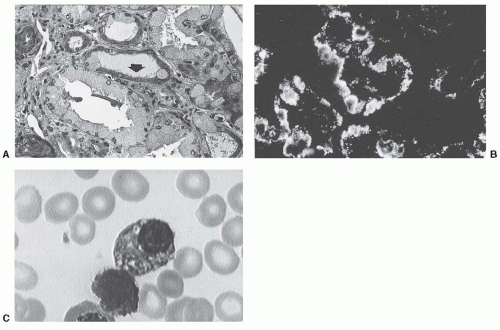
104 Proximal tubular cells are stuffed with microcrystals that stain red or green with Masson’s trichrome and are periodic acid-Schiff negative. In the most severely affected tubules, crystal-containing exfoliated cells are seen in the tubular lumen, whereas intracytoplasmic crystals are still present in atrophic tubules. In other cases, crystals can only be suspected by the presence of a finely granular material of glassy appearance in an enlarged proximal tubular epithelium (
Fig. 60.8A). Their presence is more easily demonstrated by toluidine-blue staining of semi-thin sections and by hematoxylin and eosin staining of cryostat sections. In the same tubule sections, all the cells are not equally affected; cells with a normal aspect coexist with those stuffed with crystals.
A universal feature is the additional presence of severe lesions of the proximal tubule epithelium apparently devoid of crystals. These lesions include vacuolization, loss of the luminal brush border, and focal cell sloughing, with cell fragments in the lumen of the tubules. Interstitial cellular infiltrate, including plasma cells, may contain crystalline inclusion bodies. Patchy tubular atrophy and focal interstitial fibrosis, together with a variable number of obsolescent glomeruli, are often observed.
In several cases, attempts to characterize the crystal proteins with anti-Ig conjugates, including anti-LC antibodies, have failed. When immunohistochemical studies are positive, crystals stain only (or predominantly) for the monoclonal LC, most often
κ (
Fig. 60.8B).
By electron microscopy, crystals of various size and shape (rectangular, rhomboid, round, or needle-shaped) are detected within the cytoplasm of proximal tubule cells
(
Fig. 60.8C). Intracytoplasmic crystals are surrounded by a single smooth membrane, likely of lysosomal origin.
58,
106 In rare cases, crystals are also seen in distal tubule cells. In other cases, crystals are not found by light microscopy, but electron microscopy shows enlarged vesicular bodies containing dense tubular and rod-like structures
101,
102,
103 or fibrils and needle-shaped deposits very close to crystalline structures.
Crystal formation in plasma cell dyscrasia-associated FS is not limited to renal tubule epithelium but also occurs in bone marrow and tissue-infiltrating plasma cells, and in macrophages (
Figs. 60.8C and
60.9).
103,
106 In plasma cells, crystals are localized not only in lysosomes, but they are also frequently found inside the granular endoplasmic reticulum. Crystal formation in these organelles therefore suggests incomplete proteolysis of LCs. The slow progression of myeloma disease in typical FS associated with crystal formation may be explained by the deleterious effects on cell growth of the accumulation of crystalline inclusions in the tumor plasma cells. A peculiar accumulation of LC crystals may be observed within lysosomes of macrophages in the bone marrow and other organs, defining “crystal-storing histiocytosis” (CSH). In CSH, invariably associated with κ LC monoclonal gammopathy, crystals appear to be mostly made up of monoclonal κ LC, and more rarely of entire IgG. Renal manifestations in CSH are mostly represented by chronic tubulointerstitial nephritis and FS with accumulation of monoclonal κ LC crystals within proximal tubular cells. Perirenal and interstitial infiltration by histiocytes containing eosinophilic crystalline inclusions (pseudo- pseudo Gaucher cells) is suggestive of the disease. Specific molecular peculiarities in the V domains of CSH monoclonal
κ LCs may account for crystal accumulation within histiocytes and multiple organ involvement.
113Although crystals are a salient feature of the plasma cell dyscrasia-associated FS, they are neither specific nor absolutely constant. Crystals were found in 16 of 28 (57%) patients in the two largest series published so far.
104,
105 They may also be found, albeit in low amounts, in proximal tubule epithelial cells of patients with CN,
16,
58 and occasionally in myeloma patients with isolated tubular lesions, that is, in the absence of myeloma casts.
Outcome and Treatment
As expected, patients with multiple myeloma have shorter survival time than those with MGUS. In the Mayo Clinic’s series, only one of the 14 patients with MGUS developed multiple myeloma, and at the end of follow-up, only 5 of 32 patients had evolved to end-stage renal disease.
105In patients with osteomalacia, considerable improvement can be obtained with 1
α-hydroxyvitamin D, calcium, and phosphorus supplementation. The effect of chemotherapy on the proximal tubule impairment is much more debated. It was reported that the treatment of underlying myeloma improved urinary signs and tubular transport abnormalities. However, no significant change in renal function was observed in the two largest series.
104,
105 It has been suggested that the presence of crystals within plasma cells should be added to the list of criteria against chemotherapy in
myeloma. Because chemotherapy, especially with alkylating agents, carries a significant risk of complications but without much benefit for kidney function, and because patients who do not have an overt malignancy show a relatively benign course, the risks and benefits of chemotherapy should be weighed carefully. Whether novel antimyeloma agents might improve renal prognosis in FS remains to be established.