CKD, typically when glomerular filtration rate (GFR) is <30 to 40 mL/min/m2.6,8,11,15
TABLE 77.1 Classification of Chronic Kidney Disease: Mineral and Bone Disorder | ||||||
|---|---|---|---|---|---|---|
|
inversely correlate with GFR.8,27,28,29 Patients with end-stage renal disease (ESRD) have markedly elevated levels of FGF23 that parallels with degree of hyperphosphatemia30 and secondary hyperparathyroidism (SHPT).31
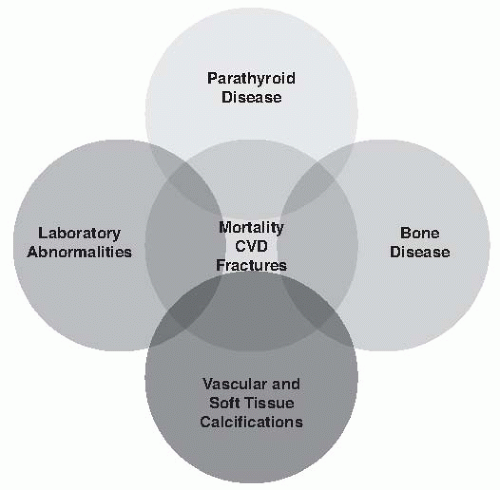 FIGURE 77.1 Spectrum of pathology in chronic kidney disease-mineral and bone disorder. (Modified from KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009;(113):S1-130.) |
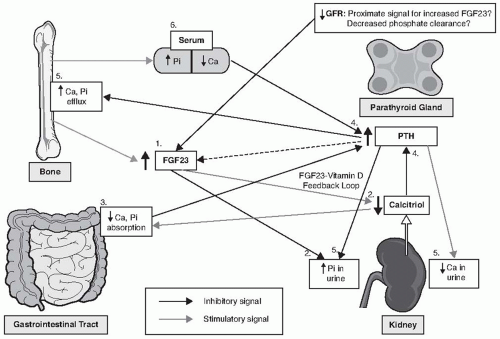 FIGURE 77.2 The schematic representation of chronic kidney disease-mineral and bone disorder pathogenesis. Ca, calcium; Pi, phosphorus. 1, Increase in FGF23; 2, suppression of calcitriol and increased urinary phosphate; 3, decreased gastrointestinal calcium and phosphorus absorption; 4, increased parathyroid hormone; 5, increased bone resorption/calcium and phosphorus bone efflux; increased phosphorus and decreased calcium in urine; 6, maintenance of serum phosphorus and calcium in normal range until stage V chronic kidney disease. |
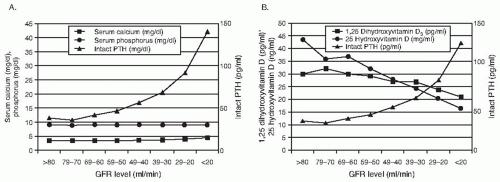 FIGURE 77.3 Relationship between glomerular filtration rate (GFR) and parathyroid hormone (PTH), calcium, phosphorus, and calcitriol levels in patients with chronic kidney disease. A: Median values of serum calcium, phosphorus, and intact PTH by GFR levels. B: Median values of 1,25-dihydroxyvitamin D,25-hydroxyvitamin D, and intact PTH by GFR levels. (Adopted from Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71(1):31-38.) |
There is increase in half-life of circulating PTH and especially C-terminal fragments observed in serum of patients with uremia, possibly due to reduced clearance as the kidney is one of the principal sites for the degradation of PTH and its fragments. Patients with advanced CKD also exhibit abnormal ratio in serum between circulating 1-84 PTH and its fragments as compared with healthy controls.58 Conventional two-site immunoassays for intact (1-84) PTH can register long N-truncated C terminal PTH fragments that lack full N terminal region (1-34) necessary for PTH1R activation. These long N-truncated C terminal fragments accumulate disproportionally to 1-84 PTH in kidney failure and may constitute up to 50% or more to total PTH immunoreactivity, as compared to 15% to 20% in normal subjects. Some of these fragments have been identified as 7-84 PTH and studies in animal models demonstrated that 7-84 PTH can antagonize effects of 1-84 PTH on increased bone turnover and serum Ca levels.59 It has been documented that patients with CKD have impaired serum Ca response to PTH and higher PTH levels are required to maintain eucalcemia. Several possible explanations of bone PTH resistance include presence of inhibitors, such as 7-84 PTH and elevated osteoprotegerin, as well as downregulation of PTH1R mRNA in animal models and patients with CKD.60,61
Although data are incomplete, the epidemiology of ROD appears to have changed in the last three decades, with a decline of OF and a higher prevalence of low bone remodeling states of uncertain clinical significance and etiology. Types of ROD also vary depending whether or not the patient already started RRT and on modality of RRT, with low turnover bone remodeling being the most common lesion in predialysis patients (27%—48%) and patients on peritoneal dialysis (48%-62%), whereas OF (32%-37%) and low turnover bone remodeling (32%—36%) occur with similar frequency in hemodialysis patients. Mixed disease represents about 10% to 13% of cases of ROD, and low turnover osteomalacia is present in 3% to 8% of patients. We lack diagnostic tools to accurately assess bone remodeling and mineralization, other than bone biopsy, making it difficult to determine the type and magnitude of bone abnormalities in an individual patient with CKD.
TABLE 77.2 Classification of Bone Disease in Chronic Kidney Disease Patients | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 77.3 TMV Classification System for Renal Osteodystrophy | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
hypogonadism, and a treatment with corticosteroids are also important clinical conditions associated with low turnover bone disease.77 There is growing evidence linking ABD to the malnutrition-inflammation complex syndrome. Higher rates of ABD are reported in peritoneal dialysis patients with low albumin levels.78 Additionally, several proinflammatory cytokines such as interleukin-1β and interleukin-6 were shown to inhibit PTH secretion in vitro.79,80 Therefore, the development of ABD is influenced by patient characteristics, as well as treatment options for CKD-MBD (Fig. 77.4).
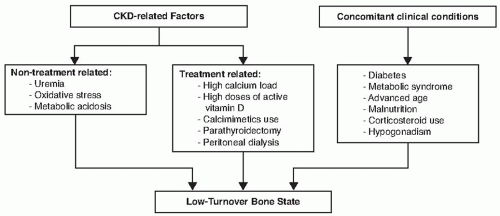 FIGURE 77.4 Low-turnover bone state risk factors. |
may influence bone formation rate independently from PTH levels. Lastly, PTH is not a bone-derived marker and therefore may never be a fully accurate indicator of bone turnover. At present, it is unknown what levels of PTH are associated with ABD in patients with less severe CKD not yet on renal replacement therapy. Bone-specific alkaline phosphatase (BSAP) may be an additional useful marker of ABD. Low levels of BSAP predict ABD and BSAP correlates with bone turnover in ESRD patients treated with hemodialysis.82
TABLE 77.4 Factors Regulating Parathyroid Hormone Secretion | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


