disorder that is truly responsible for secondarily causing MN. Evidence for secondary MN comes in situations in which treatment of the underlying process (infection, autoimmune disease, malignancy) or removal of an offending drug is temporally associated with resolution of the nephrotic syndrome, but this still does not guarantee causation because primary MN undergoes spontaneous remission in one third of cases.
TABLE 51.1 Causes of Membranous Nephropathy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
infectious disease is strengthened when the nephrotic syndrome resolves with treatment of the infection, or when antigens produced by the microorganism are consistently found within the immune deposits.
gold, and bucillamine indicate that protein excretion may continue to rise for several months after the cessation of therapy.50 The mean time to resolution of the proteinuria is 9 to 12 months, although 2 to 3 years is required in some cases.
seen by silver methenamine (Jones’ stain), which binds to basement membrane components but is not taken up by the immune deposits (Fig. 51.3). This staining, in appropriately advanced disease, reveals “spikes” of GBM present between deposits when the GBM is sectioned in cross-section, or “craters” or “pock-marks” caused by the nonsilver stained immune deposits when a tangential section of the GBM is encountered. These findings are pathognomonic for MN. The formation of immune deposits and the basement membrane response proceeds in stages according to the duration of disease (and repair). Ehrenreich and Churg (Table 51.2) classified this progression into four morphologic stages, which are more appropriate for describing the pathologic findings than correlating with clinical findings or prognosis.
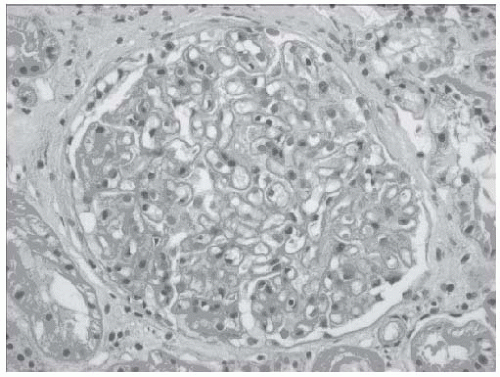 FIGURE 51.1 Hematoxylin and eosin stain (×250) of a glomerulus from a patient with idiopathic membranous nephropathy. There is diffuse thickening of the basement membrane without associated hypercellularity of the glomerular tuft. Inflammatory infiltrates are not seen and the capillary loops are widely patent. (Courtesy of Dr. Helen Cathro.) |
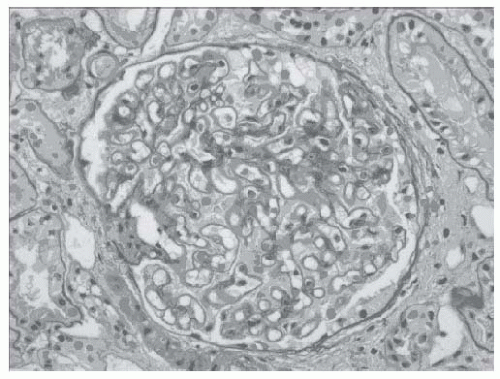 FIGURE 51.2 Periodic acid-Schiff stain of a glomerulus from a patient with idiopathic membranous nephropathy (×250). The basement membrane surrounding the capillary loops is diffusely thickened. (Courtesy of Dr. Helen Cathro.) |
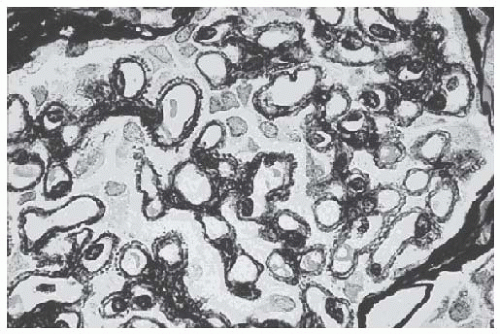 FIGURE 51.3 Jones silver stain (×250) of a glomerulus from a patient with idiopathic membranous nephropathy demonstrating “spikes” corresponding to newly synthesized basement membrane surrounding immune complexes. (Courtesy of Dr. Edward Klatt.) |
TABLE 51.2 Pathologic Staging of Membranous Nephropathy | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
primary from secondary disease, as the predominant IgG subclass in primary MN is IgG4. Secondary causes, in most cases, have a predominance of non-IgG4 subclasses, most notably in lupus-associated69,70,71 and malignancy-associated MN.49 The presence of C1q, an early component of the classical complement pathway, may also help distinguish between primary and secondary cases. Strong C1q staining is not typically found in primary MN (less than 20% of cases)69,72 but is more common in lupus-associated MN.
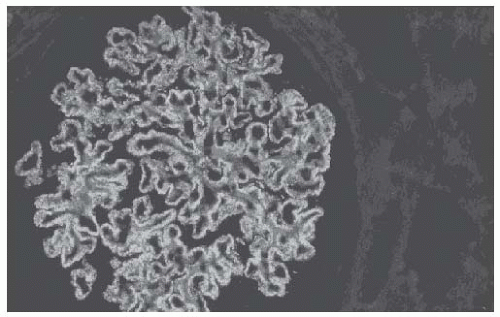 FIGURE 51.4 Immunofluorescence staining (anti-IgG) of a glomerulus from a patient with idiopathic membranous nephropathy (×250). Diffuse granular staining along the basement membrane is evident and corresponds to the deposition of immune complexes. Mesangial areas are free of immune deposits. |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


