Membranoproliferative glomerulonephritis (MPGN), sometimes referred to as mesangiocapillary glomerulonephritis, describes a pathologic pattern of injury characterized by thickening of peripheral capillary walls (membrano-), glomerular hypercellularity (-proliferative), with mesangial expansion leading to an accentuated lobular appearance of the glomerular tuft. Although there are idiopathic forms of this condition, the majority of cases of MPGN are associated with secondary causes—in particular, hepatitis C (
Table 50.1). The various forms of MPGN have very different clinical courses and are described separately.
CLASSIFICATION
Prior classifications of MPGN have subdivided this entity into three different classes according to pathologic characteristics (
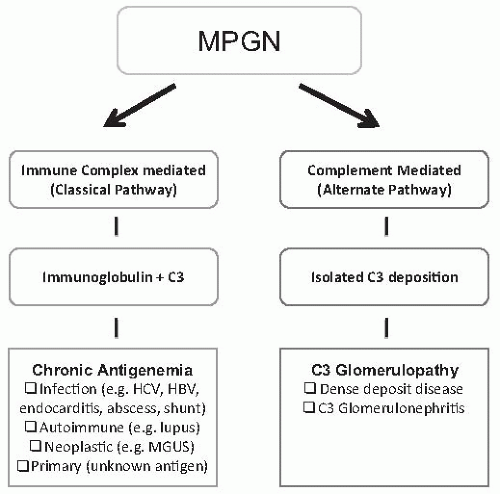
Table 1). Type I is the most common and is an immune complex mediated disorder with primarily subendothelial and mesangial immune deposits leading to activation of the classical pathway of complement. Type III was described as having a similar histologic pattern, but with the additional finding of subepithelial immune deposits or a distinctive appearance of the capillary wall deposits identified by electron microscopy. It is increasingly recognized that cases previously considered as Type III MPGN do not represent a separate disorder, but rather are best considered a variant of Type I MPGN, or more commonly, a form of the recently described entity C3 glomerulopathy. Dense deposit disease (DDD; type II) results from dysregulation of the alternate pathway of complement, and it has become clear that only a minority of cases of DDD demonstrate an MPGN pattern on biopsy. In view of these points, we prefer to classify MPGN into primary and secondary causes (
Table 50.2), and we follow an emerging consensus that DDD and C3 glomerulopathy are separate entities altogether.
1
PRIMARY MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS
Epidemiology
Primary MPGN is a relatively rare form of glomerulonephritis in developed countries, where secondary forms of MPGN are much more common. The disease is usually found in children and young adults and, interestingly, the incidence appears to be decreasing worldwide.
2,
3,
4,
5 This may be due to a true decrease in disease incidence, partly due to improved hygiene and environmental factors reducing the incidence of bacterial infections, but may also reflect the increased ability to detect secondary forms of MPGN (e.g., viral infections, monoclonal gammopathies). In developing countries, MPGN remains one of the most common forms of glomerulonephritis.
6,
7,
8 This classically has been considered secondary to the increased incidence of bacterial infections in these countries, but this has been questioned by Johnson et al., who found little evidence for active infection in cases of MPGN in Peru.
8 They have proposed that a shift in Th1/Th2 immune balance, induced by environmental and hygienic factors, may determine the incidence of a range of glomerular diseases worldwide.
9,
10Primary MPGN is more common among whites than among black or Asian populations. A genetic susceptibility is supported by the finding of an extended HLA haplotype— HLA-B8, DR3, SCO1, GLO2—more frequently in patients with MPGN I and III (13%) than in the general Caucasian population (1%), although this haplotype is not specific for MPGN.
11 Familial forms of MPGN have been described, but these are rare.
12 Some of these may be secondary to mutations encoding complement proteins or their inhibitors (discussed below).
Pathogenesis
Primary MPGN is considered to be an adaptive immune response to a chronic antigenic stimulus leading either to the subendothelial and mesangial deposition of circulating immune complexes consisting of immunoglobulin G (IgG), C3, and antigen, or deposition of circulating antigens in the glomerular capillary walls (planted antigens) which then serve as a nidus for immune complex formation in situ. In secondary forms of MPGN a wide range of antigens (primarily infectious agents) have been implicated, but in primary MPGN the causative antigen remains unknown. The binding of the Fc portion of IgG or IgM in the immune complexes
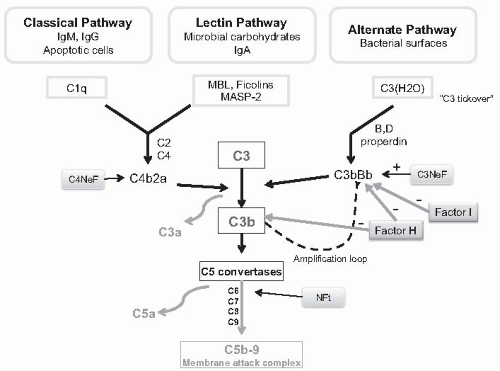
to C1q triggers activation of the complement via the classical pathway, but this initial activation may be amplified by the alternate pathway (amplification loop). The subsequent cleavage of C5 leads to the production of chemotactic factors (C5a), opsonins (C3b), and the membrane attack complex (C5b-9) (
Fig. 50.1). In some cases, complement activation is enhanced by nephritic factors that stabilize either the classical pathway C3 convertase (C4 nephritic factor, C4NeF) or the alternate pathway C3 convertase (C3 nephritic factor, C3NeF) which may enhance the amplification loop.
13,
14It is likely that the prominent monocyte influx that characterizes MPGN is the result of complex interactions including Fc portions of immunoglobulins within deposited immune complexes engaging Fc receptors on the surface of monocytes, as well as release of chemoattractant cytokines (e.g., colony stimulating factor 1 [CSF-1]) and other proinflammatory mediators by resident glomerular cells as a response to injury.
15,
16 It is unknown whether the monocytes infiltrating human glomerulonephritis (GN) have a homogeneous phenotype, and are proinflammatory, or whether some or even a majority of these cells infiltrating at different time points of the disease course may be engaged in activities primarily directed to opsonization and phagocytosis of immune complexes, stabilization of disease, and repair of glomerular injury. Recent studies suggest systemic and local innate immune responses may also contribute to the pathogenesis of MPGN. For example, studies have demonstrated toll-like receptor 3 in human MPGN associated with viral infection,
17 and upregulated expression of toll-like receptor 4 on glomerular cells has been identified in an animal model of cryoglobulinemic MPGN,
18 but more specific immune mechanisms leading to glomerulonephritis have not been established.
Following an initial proliferative phase, a second reparative phase develops, which is characterized by the development of mesangial expansion and the formation of new basement membrane matrices resulting in double contours. These duplicated basement membrane matrices lead to the sequestering of immune complexes and entrapment of cells or cell processes between the matrices (cellular interposition) and presumably this sequestration reduces the stimulation for an acute inflammatory response by circulating leukocytes.
PATHOLOGY
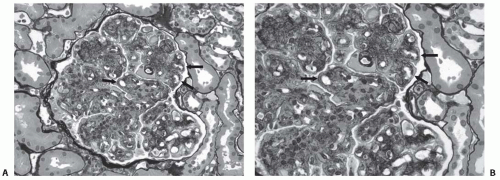
The overall appearance of both primary and secondary types of MPGN often reveals enlarged glomeruli with accentuation of the normal lobular architecture (
Fig. 50.2). Hypercellularity is mostly due to a combination of mesangial cell proliferation and infiltrating leukocytes. The mesangium is
prominently expanded, both by increased matrix as well as increased cellularity, which may reduce the number of open capillary loops. PAS or methenamine silver stains of histologic sections may show a characteristic feature of duplication (sometimes referred to as splitting) of the glomerular basement membranes (GBM) due to the synthesis of new basement membrane material, with interposition of cells (mesangial, endothelial, or leukocyte) between the duplicated basement membrane matrices.
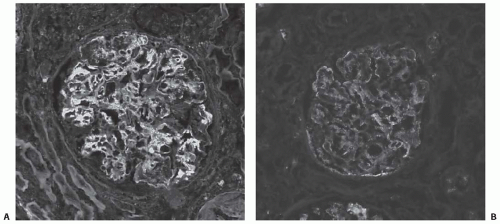
Immunofluorescence typically shows the presence of IgG and C3 in a peripheral capillary wall distribution (
Fig. 50.3). C1q and C4 are also commonly present in keeping with activation by the classical pathway. IgM is not prominent in primary MPGN and, if present, may suggest the presence of a secondary form of MPGN (e.g., hepatitis C). Glomerulonephritis with an MPGN pattern but in which deposition of immune reactants is limited to C3 has traditionally been considered part of the spectrum of MPGN I or
the former MPGN II. Recent data has suggested that many, if not all, of these cases can now be classified as DDD or manifestations of a distinct group of disorders tentatively termed C3 glomerulopathy (see later).
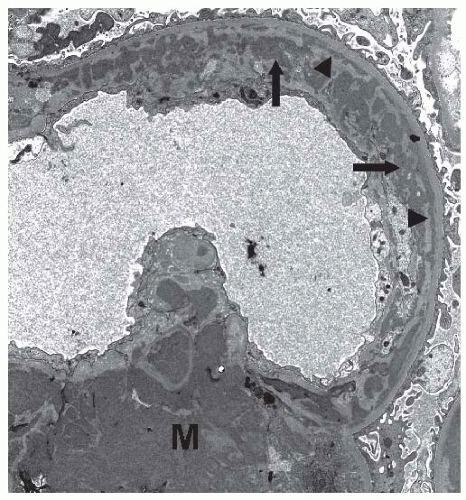
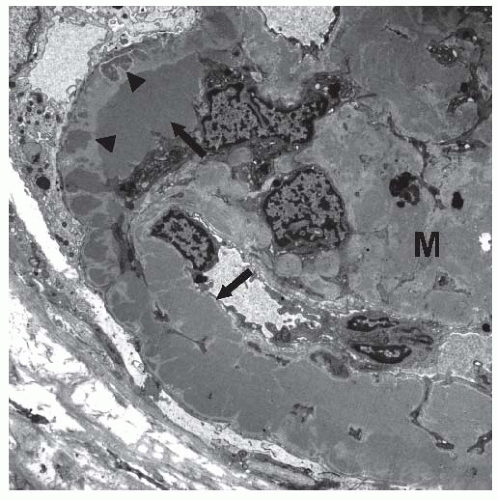
19On electron microscopy, subendothelial and mesangial deposits are typically present (
Fig. 50.4). Subepithelial deposits may be present, but are usually scanty. Prominent subepithelial deposits are found in some patients, and some authors advocate a separate classification for this entity, MPGN type III, as described later.
It is usually impossible to differentiate secondary forms of MPGN from the primary disease, but suggestive features from common forms are described in
Table 50.3. It should also be recognized that other clinical disorders may present with an MPGN pattern on renal biopsy. We have mentioned DDD, but other imitators may include thrombotic microangiopathy and other rare diseases (
Table 50.4).
Clinical Features (Presentation and Clinical Manifestations)
Type 1 MPGN often presents in childhood or early teens with edema and nephrotic syndrome, and is often associated with hematuria and an active urine sediment (dysmorphic red cells and red cell casts). Constitutional symptoms including lassitude, fatigue, and weight loss may be present.
A range of other modes of presentation may be found ranging from asymptomatic microhematuria or proteinuria on screening to an acute nephritic syndrome with gross hematuria, renal impairment, and hypertension. This is often initially diagnosed as a postinfectious glomerulonephritis, but the persistent hypocomplementemia and failure to resolve usually prompt a renal biopsy, if not already performed. In children the course is often slowly progressive, with 20% to 60% of treated patients progressing to end-stage renal disease (ESRD) over 10 to 15 years.
20,
21,
22,
23 Risk factors for progression include the presence of nephrotic syndrome and a raised serum creatinine at presentation.
In adults, primary MPGN typically presents with an overlap between nephrotic and nephritic syndrome. Nephrotic range proteinuria is common, as are microhematuria, renal impairment, and hypertension, which may be prominent. The course is variable, but in the absence of a secondary cause, a progressive course is common.
Laboratory Findings
A major clinical clue to the diagnosis of MPGN is the presence of hypocomplementemia, due to activation of the classical pathway (
Table 50.5). CH50, C3, and C4 are all depressed and may be persistent. C3 nephritic factor (C3NeF), an autoantibody stabilizing the C3 convertase of the alternate pathway, may occasionally be found (see dense deposit disease section).
13,
14 C4 nephritic factor (C4NeF), an autoantibody stabilizing the classical pathway C3 convertase or a nephritic factor of the terminal pathway (Nft), have also been described,
24 occurring in ˜20% of cases, either alone or with C3NeF.
MPGN TYPE III
MPGN Type III was originally described by Burkholder et al. in the 1970’s as a separate entity with pathological features that overlap between membranous nephropathy and a proliferative glomerulonephritis.
34 It was characterized by the presence of epimembranous “spikes” of silver staining projections of matrix from the glomerular basement membranes, in addition to the usual features of MPGN, absent C1q and C4 deposition on immunofluorescence, and by the presence of prominent sub-epithelial immune deposits with thickening and irregularity of the GBM on electron microscopy (
Figure 5). A second pattern of injury was described by the groups of Strife
35 and Anders
36, with histologic features of MPGN characterized ultrastructurally by permeation of an irregularly thickened glomerular basement membrane by electron dense deposits that are frequently poorly demarcated from the glomerular basement membrane, but are less electron dense than the deposits encountered in MPGN type I or dense deposit disease, and frequently contain areas of electron lucency. The clinical presentation of MPGN III is similar to MPGN type I, but patients tend to be older, with a more insidious presentation and hypertension is less common. Low C3 levels are typically found (˜85%), but C4 is usually normal in keeping with activation of the alternate pathway.
It is now generally considered that MPGN III is not a separate class of MPGN, and modern classifications, as recently proposed by Sethi et al.
1, omit the use of this term (
Figure 6). When immune complexes containing immunoglobulins are present, there is no compelling reason to consider MPGN III as a separate entity from MPGN I, as a distinct clinical course or outcome has not been defined, and pathologic features overlap with MPGN I. When only deposits of C3 are detected by immunofluorescence, a pattern present in most of the cases of Strife and Anders type, these cases fall into the broadly defined category of C3 glomerulopathy.
HEPATITIS C-ASSOCIATED MPGN
Hepatitis C virus (HCV) is a single-stranded RNA virus estimated to infect 170 million people worldwide. It is transmitted primarily by transfusion of blood products and intravenous drug use. Chronic infection occurs in 85% to 90% of exposed individuals and may progress to chronic active hepatitis, liver cirrhosis, and hepatocellular carcinoma. The association of HCV with cryoglobulinemic MPGN was first reported in 1993,
37 but a range of other renal diseases have also been reported with HCV infection (
Table 50.6).
Cryoglobulinemic MPGN
Cryoglobulinemia refers to the presence of serum immunoglobulins that reversibly precipitate when cooled to 4°C,
and is classified according to the composition of the circulating cryoglobulins (
Table 50.7). Type I cryoglobulins are composed of monoclonal immunoglobulins secondary to B cell disorders such as Waldenström macroglobulinemia or multiple myeloma. Type II and type III are mixed cryoglobulins consisting of polyclonal IgG complexed to another immunoglobulin which acts as an antiglobulin (anti-IgG rheumatoid factor [RF]). In type II this antiglobulin (usually IgM) is monoclonal, whereas in type III it is polyclonal. Evidence of HCV infection has been found in up to 95% of cases of type II cryoglobulinemia and 50% of type III cryoglobulinemia in some series.
38,
39 The circulating cryoglobulins are typically composed of HCV antigen and anti-HCV antibody complexed to an IgM with rheumatoid factor activity. HCV RNA is found to be concentrated 10 to 100 times in the cryoprecipitate.
37,
40The exact pathogenesis of the rheumatoid factor generation in HCV infected patients is still unknown, but may relate to B cell dysregulation following HCV infection. The E2 envelope protein of HCV has been shown to bind
directly to B lymphocytes via CD-81 receptors leading to activation and proliferation.
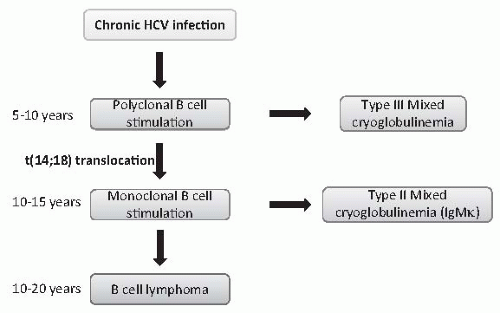
41 A range of autoantibodies are commonly found with HCV infection with an increased incidence of autoimmune diseases such as thyroid disease, diabetes, and Sjögren syndrome. It is likely that earlier in the course of HCV infection, B cell stimulation produces polyclonal IgM leading to a type III cryoglobulinemia. Over time, a B cell clone emerges, due to the acquisition of somatic genetic alterations which enhance B cell survival, leading to a monoclonal IgM antiglobulin and type II cryoglobulinemia (
Fig. 50.7). Notably, a t(14;18) translocation, which may lead to overexpression of the antiapoptotic gene
bcl-2, has been described in 80% of patients with HCV and cryoglobulinemia, but is rare (<10%) in HCV patients without circulating cryoglobulins.
42