TABLE 66.1 Effects of Diuretics on Electrolyte Excretion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
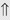 (10%-25%)
(10%-25%)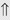 (15%-30%)
(15%-30%)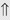 (6%)
(6%)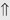 (5%-10%)
(5%-10%)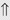 (10%-20%)
(10%-20%)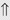 (>20%)
(>20%)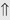 (6%)
(6%)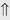 (4%)
(4%)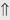 (60%)
(60%)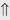 (>20%)
(>20%)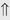 or ⇔ (<5%)
or ⇔ (<5%)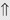 (<5%)
(<5%) (30%)
(30%) (40%)
(40%) (60%-100%)
(60%-100%) (>20%)
(>20%)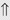 (>20%)
(>20%)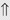 (>20%)
(>20%)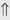 (6%-11%)
(6%-11%)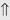 (10%)
(10%) (200%)
(200%) (>20%)
(>20%)
 (5-10%)
(5-10%)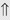 (1%-5%)
(1%-5%)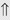 (6%)
(6%)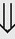 (8%)
(8%)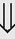
 indicates that the drug increases excretion;
indicates that the drug increases excretion;  indicates that the drug decreases excretion; ⇔ indicates that the drug has little or no direct effect on excretion. During chronic treatment, effects often wane (Na excretion), may increase (K excretion during distal convoluted tubule diuretic treatment), or may reverse as with uric acid (not shown).
indicates that the drug decreases excretion; ⇔ indicates that the drug has little or no direct effect on excretion. During chronic treatment, effects often wane (Na excretion), may increase (K excretion during distal convoluted tubule diuretic treatment), or may reverse as with uric acid (not shown).most diuretic drugs affect transport processes along several nephron segments, most owe their clinical effects primarily to their ability to inhibit Na transport by one particular nephron segment. An exception is the osmotic diuretics. Although these drugs were initially believed to inhibit solute and water flux primarily along the proximal tubule, subsequent studies have revealed effects in multiple segments. Other diuretics, however, are classified according to their primary site of action.
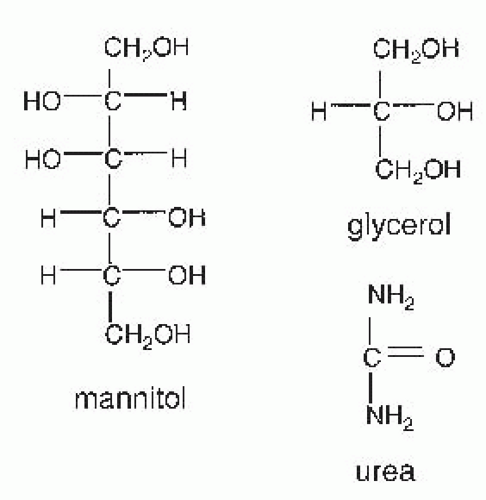 FIGURE 66.1 Structures of osmotic diuretics. |
inhibitors have a limited therapeutic role as diuretic agents, however, because they are only weakly natriuretic when employed chronically. They are used primarily to reduce intraocular pressure in glaucoma, to enhance bicarbonate excretion in metabolic alkalosis or chronic hypercapnia, and to prevent mountain sickness. Structures of carbonic anhydrase inhibitors are shown in Figure 66.2.
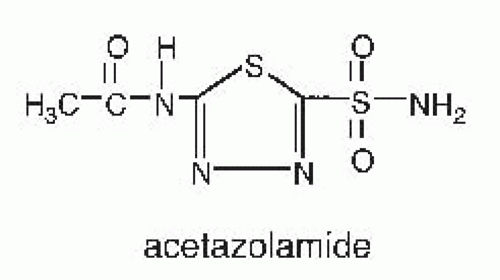 FIGURE 66.2 Structure of a carbonic anhydrase inhibitor. |
tubules reabsorb bicarbonate is depicted in Figure 66.3. The effect of carbonic anhydrase to accelerate bicarbonate is a result of the reactions that occur in both luminal fluid and in the cell. The mechanism of carbonic anhydrase action in luminal fluid,52 is shown here, where E represents the carbonic anhydrase enzyme:
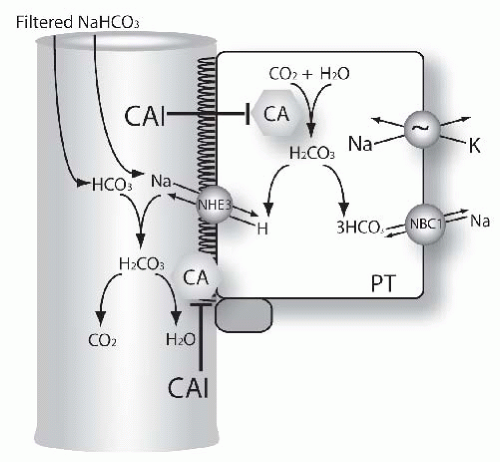 FIGURE 66.3 Mechanisms of diuretic action in the proximal tubule. The figure shows functional model of proximal tubule (PT) cells. Many transport proteins are omitted from the model, for clarity. Carbonic anhydrase (CA) catalyzes inside the cell the formation of HCO3 from H2O and CO2. This is the result of the two-step process (please see equations in the text for additional details). Bicarbonate leaves the cell via the Na, HCO3, cotransporter.186,187 A second pool of carbonic anhydrase is located in the brush border (CA). This participates in disposing of carbonic acid, formed from filtered bicarbonate and secreted H+. Both pools of carbonic anhydrase are inhibited by acetazolamide and other carbonic anhydrase inhibitors (see text for details). |
tubular sites limits net fractional excretion of bicarbonate to ˜25% to 30%, even during acute administration.56,57
an alkaline urine and hypercalciuria.73 Other adverse effects include drowsiness, fatigue, central nervous system depression, and paresthesias. Bone marrow suppression has been reported.74,75
contrasts with that of osmotic diuretics which increase osmolar clearance and CH2O.97 During hydropenia, loop diuretics impair the reabsorption of solute-free water (
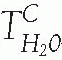 ). During maximal loop diuretic action, the urinary Na concentration is usually between 75 to 100 mM.98 Because urinary K concentrations during furosemide-induced natriuresis remain low, this means that the clearance of electrolyte free water (CH2Oe) is increased when loop diuretics are administered during conditions of water diuresis or hydropenia.98 This effect of loop diuretics has been exploited to treat hyponatremia, when combined with normal or hypertonic saline.99,100
). During maximal loop diuretic action, the urinary Na concentration is usually between 75 to 100 mM.98 Because urinary K concentrations during furosemide-induced natriuresis remain low, this means that the clearance of electrolyte free water (CH2Oe) is increased when loop diuretics are administered during conditions of water diuresis or hydropenia.98 This effect of loop diuretics has been exploited to treat hyponatremia, when combined with normal or hypertonic saline.99,100 FIGURE 66.4 Structures of loop diuretics. |
perhaps via inhibition of prostaglandin dehydrogenase.111,112 Blockade of cyclooxygenase reduces the effects of furosemide to inhibit loop segment chloride transport in rats,113,114 and this effect appears to be important clinically because nonsteroidal anti-inflammatory drugs (NSAIDs) are common causes of diuretic resistance (see below). Increases in renal prostaglandins may also contribute to the hemodynamic effects of loop diuretics, described later.
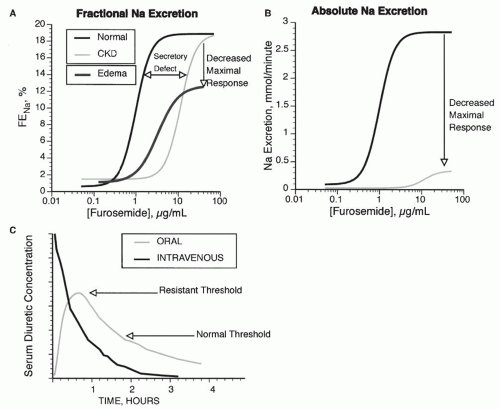 FIGURE 66.5 Dose response curve for loop diuretics. A: The fractional Na excretion (FENa) as a function of loop diuretic concentration. Compared with normal patients, patients with chronic renal failure (CKD) show a rightward shift in the curve, owing to impaired diuretic secretion. The maximal response is preserved when expressed as FENa, but when expressed as absolute Na excretion (B), maximal natriuresis is reduced in patients with CKD. Patients with edema demonstrate a rightward and downward shift, even when expressed as FENa (A). C: Compares the response to intravenous and oral doses of loop diuretics. In a normal individual (Normal), an oral dose may be as effective as an intravenous dose because the time above the natriuretic threshold (indicated by the normal line) is approximately equal. If the natriuretic threshold increases (as indicated by the dashed line, from an edematous patient), then the oral dose may not provide a high enough serum level to elicit natriuresis. |
the interstitium, drives calcium and magnesium absorption through the paracellular pathway. Loop diuretics, by blocking the activity of the Na-K-2Cl cotransporter at the apical membrane of thick ascending limb cells, reduce the transepithelial voltage toward or to 0 mV. This stops passive paracellular calcium and magnesium absorption.
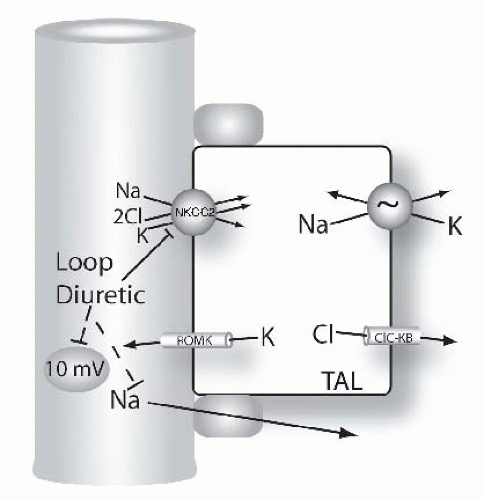 FIGURE 66.6 Mechanisms of diuretic action along the loop of Henle. Figure shows model of thick ascending limb (TAL) cells. Na and Cl are reabsorbed across the apical membrane via the loop diuretic-sensitive Na-K-2Cl cotransporter, NKCC2. Loop diuretics bind to and block this pathway directly. Note that the transepithelial voltage along the TAL is oriented with the lumen positive relative to blood (circled value, given in millivolts, mV). This transepithelial voltage drives a component of Na (and calcium and magnesium, see Fig. 66.9) reabsorption via the paracellular pathway. This component of Na absorption is also reduced by loop diuretics because they reduce the transepithelial voltage. |
RBF under experimental conditions.138,139 This effect is believed related to the diuretic-induced production of vasodilatory prostaglandins (discussed previously).
Interestingly, the response to loop diuretics is also associated with polymorphisms in the genes encoding the more distal sodium transporters NCC and ENaC.162
TABLE 66.2 Pharmacokinetics of Loop Diuretics | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Immunobiology and Immunopharmacology of Renal Allograft Rejection
Immunobiology and Immunopharmacology of Renal Allograft Rejection
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


