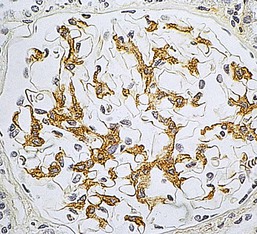
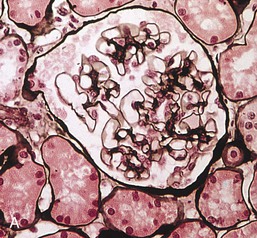
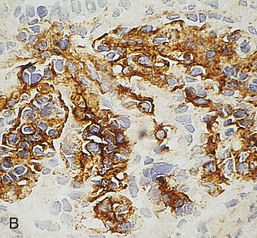
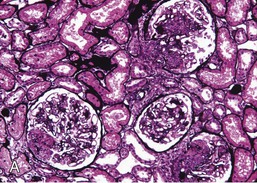
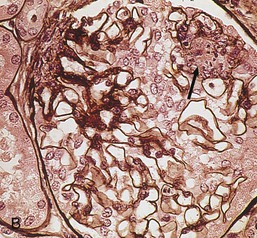
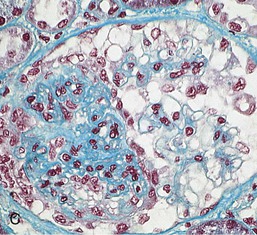
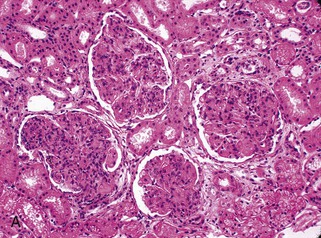
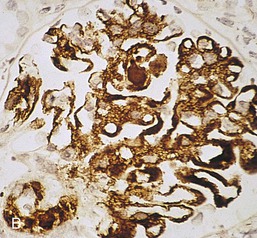
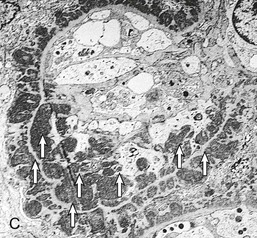
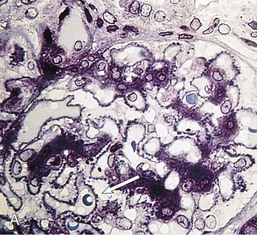
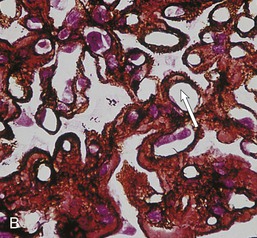
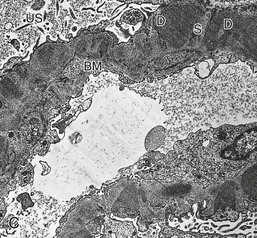
Gerald B. Appel, David Jayne, Brad H. Rovin Lupus nephritis (LN) is an immune complex glomerulonephritis that is a common and serious feature of systemic lupus erythematosus (SLE), which itself is defined by a combination of clinical and laboratory features.1,2 The American College of Rheumatology (ACR) criteria for the diagnosis of SLE have been widely used in both epidemiologic and treatment studies (Box 26-1). However, many patients with “lupus-like” conditions and others who meet fewer than four ACR criteria should still be recognized as requiring therapy. The incidence and prevalence of lupus and LN are influenced by age, gender, ethnicity, geographic region, diagnostic criteria used, and method of ascertainment, but across populations, clinically important kidney disease will occur in 40% of patients3–7 (Table 26-1). The peak incidence of lupus is age 15 to 45 years, with women outnumbering men by 10 : 1. This gender predominance is less pronounced in children and older individuals. Among lupus patients, LN affects both genders equally and is more severe in children and men and less so in older adults. Both lupus and LN are three to four times more common in blacks, Asians, and Hispanics than in Caucasians. Additional risk factors for LN include younger age, lower socioeconomic status, more ACR criteria for SLE, longer disease duration, family history of SLE, and hypertension. Multiple genes predispose to lupus.8 This is supported by the clustering in families, racial differences in susceptibility, concordance of SLE in more than 25% of identical twins but only in 5% of fraternal twins, and frequency of positive autoantibodies and other autoimmune disorders in family members of SLE patients. Homozygous deficiency of complement components (C1q, C2, C4) carries a high risk for development of SLE; this is also true for certain FcγRIII receptor polymorphisms. Genome-wide association studies have identified 17 loci associated with an increased risk of SLE that involve genes associated with B cell signaling, Toll-like receptors, and neutrophil function.8 Environmental factors also play a role in the onset and exacerbation of SLE and LN.9 Hormonal factors are suggested by the strong female predisposition, the exacerbations during or shortly after pregnancy, and the role of hormonal treatment and ablation in animal models of LN. Whereas exposure to certain medications can produce SLE or a lupus-like syndrome, LN occurs infrequently in these patients. Conclusive evidence for a viral pathogenesis of SLE or LN has yet to be produced. Spontaneous and inducible models of SLE in mice include the NZB B/W F1 hybrid, the BXSB, and the MRL/lpr mice. Some of these models have defective apoptosis that lead to defective clonal deletion and B cell proliferation. SLE can be induced in animals by injection of autoantibodies against DNA, phospholipids, or peptides derived from the Smith antigen. Patients with SLE typically develop a wide range of autoantibodies.10 Early stages of disease evolution include the defective clearance of apoptotic cells and release of autoantigens from these cells, stimulating interferon-α (IFN-α) and proinflammatory cytokine production. This results in a breakdown of self-tolerance, autoantibody production, and immune complex deposition. Many autoantibodies are directed against nucleic acids and proteins concerned with transcriptional and translational machinery, such as nucleosomes (DNA-histone), chromatin antigens, and small nuclear and cytoplasmic ribonuclear proteins. Polyclonal hyperactivity of B cells along with defective autoregulation of T cells is thought to underlie autoantibody production.10,11 A number of mechanisms may contribute to SLE, including failure to remove or to silence autoreactive B and T cells, abnormal exposure to or presentation of self antigens, T cell hyperactivity, increased B cell–stimulating cytokines, and B cell hyperactivity.10 The failure of apoptotic mechanisms to delete or to silence autoreactive cells (tolerance) may allow clonal expansion of such cells later in life, leading to autoantibody production. Abnormal exposure to self antigen may occur through nuclear autoantigens clustering on the blebs of apoptotic cells, which may be associated with germ-line mutations that lead to clonal expansion of autoreactive cells. Likewise, “antigen mimicry” may occur if exposure to viral or bacterial peptides containing sequences similar to native antigens leads to a similar induction of autoantibody-producing cell lines. The nature of antigen presentation may also be important, with certain nuclear antigens capable of triggering an immunogenic response through interactions with a variety of intracellular Toll-like receptors. In SLE, there is evidence that T cell hyperactivity and failure of T cell tolerance may allow autoreactive T cells to drive B cell proliferation and the production of autoantibodies. Also, proliferation of autoreactive B cells may occur through a variety of disturbances of positive and negative regulatory mechanisms (e.g., stimulation by superantigens) in addition to the mechanisms mentioned. The end result is the loss of tolerance and production of a wide array of autoantibodies.10,11 Autoantibodies are crucial to the pathogenesis of both SLE and LN. In SLE, some autoantibodies, such as those causing autoimmune hemolytic anemia, are directly pathogenic. Other autoantibodies combine with antigen to produce immune complexes that if not adequately cleared may deposit in various organs, inciting inflammatory responses.1 Complement components aid apoptotic clearance and removal of immune complexes, but they are also activated by immune complexes and contribute to the inflammatory cascade. A hallmark of glomerular involvement in LN is the accumulation of immune complexes. Patients with LN have autoantibodies against double-stranded DNA (dsDNA), Sm antigen, C1q, nucleosomes, and other antigens, and there is evidence both for direct binding of anti-DNA antibodies to the glomerular basement membrane (GBM) and for cross-linking of positively charged nucleosome components such as chromatin between autoantibodies and GBM.12–14 In proliferative LN, DNA and high-affinity complement-fixing antibodies are found in the subendothelial space, and this may be facilitated by cationic histones binding to the negatively charged GBM. In general, mesangial and subendothelial immune deposits are derived from deposition of circulating immune complexes, whereas subepithelial immune complexes may include those formed in situ. However, whether deposition of circulating complexes or in situ formation is the predominant pathogenic pathway in a given patient is not always clear. The localization of immune complexes within the glomerulus is influenced by size, charge, specificity, and avidity as well as by the clearing ability of the mesangium and local hemodynamics.12–16 The localization of immune complexes in the glomerulus leads to complement activation and complement-mediated damage, activation of procoagulant factors, leukocyte Fc receptor activation driving leukocyte infiltration into the kidneys with release of their proteolytic enzymes, and activation of cytokines associated with cellular proliferation and matrix formation. Nucleosomes can also activate resident dendritic cells (see later) through binding to Toll-like receptors 2 and 9. Intraglomerular hypertension and activation of coagulation cascades may contribute to the glomerular injury, especially in patients with antiphospholipid antibodies (aPLAs, APA). The obligatory role of autoantibodies and immune complexes in the pathogenesis of LN has been challenged. In a mouse model of SLE in which B cell secretion of antibodies is prevented, LN still developed, although the glomerular histology was not as inflammatory as in wild-type mice.17 Also renal biopsies from lupus patients without clinical LN still show immune deposits.18,19 It is thus unclear why all patients with SLE do not develop LN, and this suggests renal immune complexes may be necessary but not sufficient for the development of the full clinical phenotypes of LN. T cell subsets appear to contribute to the progression of LN. T helper type 17 (Th17) cells have been observed in human renal biopsies, and by producing interleukin-17 (IL-17) locally, may drive the expression of inflammatory cytokines and chemokines by renal parenchymal cells, recruiting and activating neutrophils and monocytes. Th17 cells in the kidney may reflect a shift away from T regulatory cells (CD4+CD25hiFoxP3+), which inhibit immune responses and autoantibody production.20 There has been increasing recognition of the role of neutrophils in SLE and LN. Neutrophils undergo a novel form of cell death called NETosis in which a chromatin meshwork (a NET) is released.21 These NETs (neutrophil nets) are a source of autoantigens and are not degraded properly in lupus patients. In addition, lupus patients have an increased number of low-density granulocytes that are more susceptible to NETosis. NET material has been found in LN biopsies, and SLE patients who are nondegraders often had LN.22,23 The NETs induce production of IFN-α by plasmacytoid dendritic cells, which are also found in LN kidneys.24,25 IFN-α appears to have a central role in SLE and LN. IFN-α drives dendritic cell maturation to antigen-presenting cells, B cell differentiation to antibody-producing plasma cells, and development of effector and memory T cells.26–29 Within the kidney interstitium, T and B cells may associate and in some cases develop germinal centers.30 These B cells show clonal expansion and somatic hypermutation, suggesting intrarenal autoantibody production, potentially against specific renal antigens. These interactions likely contribute to interstitial inflammation in LN, which is a major determinant of long-term renal survival.30 Systemic lupus erythematosus can affect virtually any organ of the body. The disease course is characterized by episodes of illness (flares) followed by periods of relative quiescence (remissions). Nonhealing damage is a consequence of inflammation and is a strong predictor of outcome. A number of reliable and reproducible scoring systems have been devised to follow the activity and development of damage of an individual patient with SLE. These include the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI), the British Isles Lupus Assessment Group (BILAG), the Systemic Lupus Activity Measure (SLAM), and the Systemic Lupus International Collaborating Clinic–American College of Rheumatology (SLICC-ACR) damage score. SLE often results in profound disturbance of quality of life and disruption of economic and reproductive activity. Renal involvement is usually manifested by proteinuria, active urinary sediment with microhematuria, dysmorphic erythrocytes and erythrocyte casts, and hypertension. In many cases with major renal involvement, the nephritic syndrome develops in association with proliferative glomerulonephritis (GN) and a decline in glomerular filtration rate (GFR). Clinical findings correlate well with histologic glomerular findings (see later discussion). Infrequently, renal disease in SLE presents with tubular disorders such as distal renal tubular acidosis with hypokalemia (type 1 RTA) or hyperkalemia (type 4 RTA) (see Chapter 12), thrombotic disorders associated with a secondary antiphospholipid antibody syndrome (see later and Chapter 28), and fibrillary GN (see Chapter 27). Patients with active SLE often present with nonspecific complaints of malaise, low-grade fever, poor appetite, and weight loss. Other common features include patchy alopecia; oral or nasal ulcerations; arthralgias and nondeforming arthritis; and a variety of dermal findings, including photosensitivity, Raynaud’s phenomenon, and the classic “butterfly” facial rash. Livedo reticularis is seen in up to 15% of cases and may be associated with miscarriages, thrombocytopenia, and APA.31 Neuropsychiatric involvement presents with headache, nerve palsies, frank coma, and psychoses. Serositis, in the form of pleuritis or pericarditis, affects up to 40% of patients. Pulmonary hypertension can develop silently as a result of multiple pulmonary emboli or intravascular coagulation in association with APA or may be caused by nonthrombotic pulmonary arterial disease. Libman-Sacks endocarditis and much more common mitral valve prolapse can be detected with clinical findings or by echocardiography. Splenomegaly and lymphadenopathy are present in about one fourth of patients. Hematologic abnormalities in SLE include anemia caused by impaired erythropoiesis, autoimmune hemolysis, and bleeding. Thrombocytopenia and leukopenia may be part of the disease process or may result from complications of therapy. Thrombotic events should prompt a search for APA and other procoagulant abnormalities. Whereas the diagnosis of lupus may be obvious in a young female with classic manifestations and serologic markers, less typical presentations can result in multiple physician consultations and diagnostic delay. This is partly a result of the varied features of the disease and the evolution of signs and symptoms over time. Some patients, especially those with membranous LN, may present with renal disease as their initial manifestation. The presence of four or more ACR criteria carries a 96% sensitivity and specificity for lupus (see Box 26-1). However, the ACR diagnostic criteria were developed for clinical studies and do not always prove useful in an individual patient. Other diseases mimicking SLE include fibromyalgia, Sjögren’s syndrome, hematologic diseases such as the thrombotic microangiopathies, primary antiphospholipid syndrome, dermatomyositis, and systemic sclerosis (see Chapters 28 and 29). However, SLE may also present with overlapping features of other multisystem or organ-limited autoimmune syndromes. Nephritis has been reported in patients with mixed connective tissue disease associated with the presence of anti-Ro and anti-La antibodies and the absence of anti-dsDNA antibodies. Rheumatoid arthritis can be associated with mesangial proliferative glomerulonephritis or renal disease due to AA (secondary) amyloidosis. Older lupus patients may present with joint deformities typical of rheumatoid arthritis. Several common forms of glomerulonephritis must be distinguished from LN, since they may have some clinical features in common. These include Henoch-Schönlein purpura, antineutrophil cytoplasmic antibody (ANCA)–associated glomerulonephritis (see Chapter 24), bacterial endocarditis, and cryoglobulinemia. ANCAs of uncertain significance are detected in 20% of LN patients. Antinuclear antibody (ANA), found in more than 90% of untreated patients, is a highly sensitive screen for SLE patients; however, ANAs are nonspecific and found in other rheumatologic and nonrheumatologic diseases as well. Some lupus-like patients with negative ANA test results have APA, and one third of SLE patients will have APA.32–34 The pattern of ANA (e.g., diffuse, speckled) is not reliable in distinguishing lupus from other rheumatologic diseases. Autoantibodies against dsDNA are more specific, being present in 75% of untreated lupus patients, but are a less sensitive marker. Whereas high titers of anti-dsDNA antibodies correlate with the presence of SLE and are often used to follow the course of LN, antibodies to single-stranded DNA (ssDNA) are found in many rheumatologic conditions and do not correlate with the course of LN. Sm antibodies are strongly associated with the diagnosis of lupus and the presence of nephritis but are present in only about 25% to 30% of patients. Antibodies to C1q (anti-C1q) have been more closely associated with the activity of LN than anti-dsDNA antibodies and may have a prognostic role in the follow-up of patients with LN.35 Serum levels of total hemolytic complement and complement components C3 and C4 are often depressed in untreated SLE and especially LN patients. Either both C3 and C4 are depressed or C4 is preferentially depressed in lupus patients, usually reflecting preferential activation of the classical complement pathway. In patients with postinfectious glomerulonephritis (PIGN), C3 glomerulopathies and some idiopathic membranoproliferative GN, C3 is preferentially depressed (see Chapters 21 and 22). Alternatively, low C4 with normal C3 may reflect genetic C4 deficiency in lupus patients or the presence of cryoglobulins. Although LN may affect all structures of the kidney, glomerular involvement has been the best studied component and correlates well with the presentation, course, and treatment. The systematic classification systems for LN are based on glomerular involvement. The currently used International Society of Nephrology (ISN)/Renal Pathology Society (RPS) classification refines and clarifies some of the deficiencies of the previous WHO classification,36 and has improved interobserver reproducibility and the predictive value of LN biopsies37,38 (Table 26-2). Table 26-2 Classification of lupus nephritis (LN) using the ISN/RPS classification (2004). In the International Society of Nephrology/Renal Pathology Society (ISN/RPS) classification, the distribution of hypercellularity is assessed as mesangial, endocapillary, or extracapillary (crescentic) and as focal (with <50% glomerular involvement) versus diffuse (with ≥50% glomeruli affected). The distribution of immune deposits by immunofluorescence (IF) and electron microscopy (EM) is judged as mesangial, subepithelial, or subendothelial; LM, light microscopy. Class I of the ISN/RPS classification for LN denotes normal glomeruli by light microscopy but with mesangial immune deposits (Fig. 26-1). All higher classes of LN show mesangial immune deposits in addition to their specific differentiating features. Class II, mesangial proliferative LN, is defined as pure mesangial hypercellularity (more than three mesangial cells in areas away from the vascular pole in 3-µm-thick histologic sections) by light microscopy with mesangial immune deposits (Fig. 26-2). Class III, focal LN, is defined as focal segmental or global endocapillary or extracapillary glomerulonephritis affecting less than 50% of the total glomeruli sampled (Fig. 26-3). Class IV, diffuse LN, has diffuse segmental or global endocapillary or extracapillary GN affecting 50% or more of glomeruli (Fig. 26-4). Both class III and class IV have subendothelial immune deposits. Class IV is subdivided into diffuse segmental (IV-S) proliferation (i.e., >50% of affected glomeruli have segmental lesions) and diffuse global (IV-G) proliferation (i.e., >50% of affected glomeruli have global lesions). Both class III and class IV may have active (proliferative), inactive (sclerosing), or combined active and inactive lesions, subclassified as A, C, and A/C, respectively. Class V, membranous LN, is defined by subepithelial immune deposits (Fig. 26-5). The membranous alterations may be present alone or on a background of mesangial hypercellularity and mesangial immune deposits. Patients with additional true focal or diffuse proliferative lesions and subendothelial immune complex deposits are classified as V + III and V + IV under the ISN/RPS classification (see Table 26-2). Class VI, advanced sclerosing LN, is defined by global glomerular sclerosis affecting 90% or more of glomeruli. On immunofluorescence (IF), IgG is almost always the dominant immunoglobulin, and early complement components such as C4 and especially C1q are usually present along with C3. The presence of all three immunoglobulins, IgG, IgA, and IgM, along with the two complement components, C1q and C3, so called “full-house” staining, is highly suggestive of LN, as is strong C1q staining. Fibrin is often present in the glomerular tuft and especially in crescents. On electron microscopy (EM) the distribution of immune deposits corresponds to that of IF. Some electron-dense deposits have an organized substructure known as fingerprinting, corresponding to the presence of curvilinear microtubular or fibrillar structures composed of bands ranging from 10 to 15 nm in diameter. Tubuloreticular inclusions, 24-nm interanastomosing tubular structures located in the dilated cisternae of endoplasmic reticulum of renal endothelial cells, are often found in biopsy specimens of LN patients and are thought to reflect increased interferon expression. Although LN classification is based on the degree of glomerular involvement, histologic and clinical involvement of other renal compartments is seen in lupus patients.39,40 This is a deficit of the ISN/RPS system, because interstitial and vascular injury are predictors of kidney outcomes.41 In about 50% of patients with LN, predominantly those with proliferative glomerular lesions, immune aggregates are found along the tubular basement membranes. Interstitial infiltrates of CD4+ and CD8+ T lymphocytes and monocytes are frequently found. More than half of patients have significant T cell–B cell aggregates in the renal interstitium, and a small percentage have typical-appearing germinal centers.30 In active disease, infiltration and invasion of the tubules (tubulitis) is found (Fig. 26-6). In chronic disease, the interstitium is expanded by fibrosis and sparser infiltrates. Interstitial inflammation has been correlated with renal dysfunction and hypertension, whereas immune deposition along the tubular basement membranes correlates better with the presence of high anti-dsDNA and depressed serum complement levels. Infrequently, tubulointerstitial nephritis is seen in the absence of glomerular disease and may produce acute kidney injury or RTA.
Lupus Nephritis
Definition
Epidemiology
Etiology and Pathogenesis
Genetics, Environment, and Animal Models
Autoimmunity in Systemic Lupus Erythematosus
Pathogenesis of Lupus Nephritis
Clinical Manifestations
Renal Manifestations
Extrarenal Manifestations
Diagnosis and Differential Diagnosis
Immunologic Tests in Lupus
Pathology
ISN/RPS Classification of Lupus Nephritis
ISN/RPS (2004)
Class
Definition
I
Minimal mesangial LN
Normal glomeruli by LM, but mesangial immune deposits by IF
II
Mesangial proliferative LN
Mesangial hypercellularity with mesangial immune deposits
III
Focal LN
III (A): Purely active lesions: focal proliferative LN
III (A/C): Active and chronic lesions: focal proliferative and sclerosing LN
III (C): Chronic inactive lesions with glomerular scars: focal sclerosing LN
IV
Diffuse LN
IV-S (A) or IV-G (A): Purely active lesions: diffuse segmental (S) or global (G) proliferative LN
IV-S (A/C) or IV-G (A/C): Active and chronic lesions: diffuse segmental or global proliferative and sclerosing LN
IV-S (C) or IV-G (C): Inactive with glomerular scars: diffuse segmental or global sclerosing LN
V
Membranous LN
VI
Advanced sclerosing LN
≥90% of glomeruli globally sclerosed without residual activity
Classification of Lupus Nephritis
Tubulointerstitial and Vascular Disease
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Lupus Nephritis
Chapter 26