cirrhosis, studies in experimental animals have provided conclusive evidence indicating that sodium retention precedes ascites formation, further emphasizing the important role of this abnormality of renal function in the pathogenesis of ascites in cirrhosis.28,29,30,31,32 This observation suggests that sodium retention is the cause and not the consequence of ascites formation in cirrhosis.
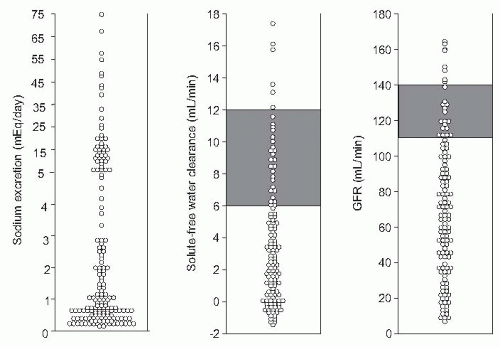 FIGURE 68.1 Individual values of sodium excretion, solute-free water clearance, and glomerular filtration rate in a large series of patients with cirrhosis and ascites without diuretic therapy and under a low-sodium diet. Lines indicate normal ranges. For urine sodium normal range is 80 to 100 mEq per day. |
in the proximal tubule, suggest that cirrhotic patients with ascites show a marked increase in proximal sodium reabsorption.46,47 Nevertheless, distal sodium reabsorption is also increased, especially in patients with more avid sodium retention.47 Clinical studies using spironolactone to antagonize the mineralocorticoid receptor indicate that this agent induces natriuresis in a large proportion of cirrhotic patients with ascites without renal failure, which supports a major role for increased sodium reabsorption in distal sites of the nephron in these patients.36,48,49,50,51 Taken together, these results suggest that in patients with cirrhosis without renal failure, an enhanced reabsorption of sodium in both proximal and distal tubules contributes to sodium retention. Potential mediators of this increased sodium reabsorption include changes in the hydrostatic and colloidosmotic pressures in the peritubular capillaries and increased activity of the sympathetic nervous system and the renin-angiotensin-aldosterone system (RAAS). Sodium retention is usually more marked in patients with renal failure than in those without renal failure due to both a reduction in filtered sodium load and a more marked activation of sodium-retaining mechanisms.
(i.e., urine sodium <10 mEq per day) in whom a positive sodium balance is anticipated despite a restriction in sodium intake should be started on moderately high doses of aldosterone antagonists (e.g., spironolactone 100 to 200 mg per day) alone or in association with loop diuretics (e.g., furosemide 40 mg per day). Conversely, patients with moderate sodium retention (i.e., urine sodium > 10 mEq per day) would likely respond to low doses of aldosterone antagonists (i.e., spironolactone 25 to 100 mg per day). The use of higher doses of spironolactone in these latter patients may induce overdiuresis and cause dehydration, hypovolemic hyponatremia, and prerenal renal failure. Besides its importance in helping establish the dose of diuretics, the intensity of sodium retention also provides prognostic information in patients with ascites. Patients with baseline urine sodium lower than 10 mEq per day have a median survival time of only 1.5 years compared with 4.5 years in patients with urine sodium higher than 10 mEq per day (Fig. 68.2).60,61,62 Finally, the measurement of sodium excretion in patients under diuretic therapy is very useful to monitor the response to treatment.
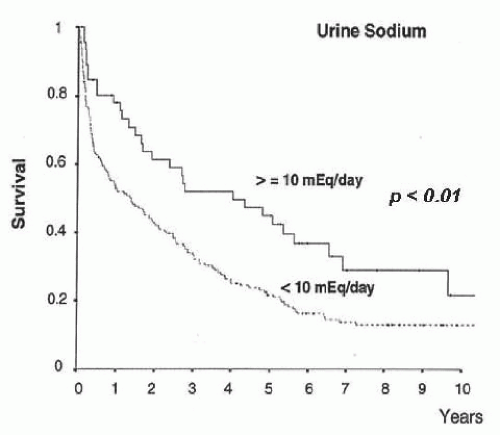 FIGURE 68.2 Long-term survival according to sodium excretion in a series of 204 patients with cirrhosis admitted to the hospital for the treatment of ascites. |
ascending limb of the loop of Henle, reduced renal synthesis of prostaglandins, and nonosmotic hypersecretion of arginine vasopressin (AVP).68,85,86,87 Although definitive data about the relative importance of these factors in the pathogenesis of hyponatremia in patients with cirrhosis is lacking, it is likely that AVP hypersecretion plays a major role. This contention is supported by studies in animals and patients with cirrhosis showing that the administration of vaptans, drugs that antagonize the tubular effects of AVP (V2 receptor antagonists), improve solute-free water excretion and increase serum sodium concentration.88,89,90,91,92,93,94,95 However, it is important to note there is a significant number of patients in whom hyponatremia does not improve despite the administration of vaptans, thus suggesting that factors other than AVP play also a role in the pathogenesis of solute-free water retention in cirrhosis. In patients with renal failure it is likely that besides AVP, a reduced distal delivery of filtrate due to decreased filtered load and increased proximal sodium and water reabsorption plays a role in solute-free water retention.
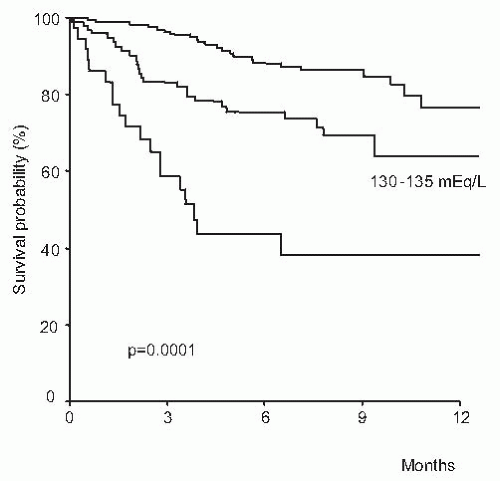 FIGURE 68.3 One-year survival before transplantation in a series of 308 patients with cirrhosis according to different values of serum sodium. (Reproduced with permission from Londoño MC, Cárdenas A, Guevara M, et al. MELD score and serum sodium in the prediction of survival of patients with cirrhosis awaiting liver transplantation. Gut. 2007;56:1283-1290.) |
would probably not tolerate a further challenge to cell volume, and encephalopathy would develop due to any other osmotic stimulus, including situations associated with an increased ammonia load to the brain (gastrointestinal hemorrhage, infection) or further impairment in serum sodium concentration (Fig. 68.4). Several lines of evidence support the existence of a relationship between hepatic encephalopathy and low serum sodium concentration. First, serum sodium levels and serum ammonia levels are major factors determining electroencephalographic abnormalities in cirrhosis.101 Second, in patients treated with transjugular intrahepatic portosystemic shunts, hyponatremia is a major risk factor for hepatic encephalopathy.102 Third, in patients treated with diuretics (a clinical situation associated with a high incidence of hepatic encephalopathy), hyponatremia is a risk factor for hepatic encephalopathy (P. Ginès, unpublished data). Finally, serum sodium has been shown to be an independent predictive factor of hepatic encephalopathy in several series of patients with advanced cirrhosis.103,104,105
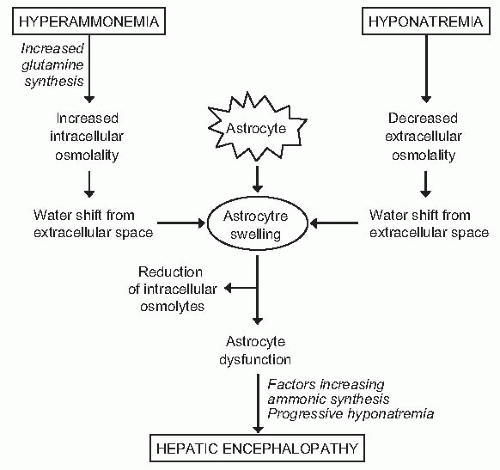 FIGURE 68.4 Proposed interaction between hyperammonemia and hyponatremia on brain astrocytes and possible pathogenic relationship with hepatic encephalopathy. (Reproduced with permission from Ginès P, Guevara M. Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management. Hepatology. 2008;48:1002-1010.) |
be triggered by some precipitating factors, particularly bacterial infections.119,120,121 When renal perfusion is estimated by sensitive clearance techniques, such as para-aminohippurate or inulin clearances, in a population of hospitalized patients with ascites, normal values are found in only one fifth of cases. In another 15% to 20%, renal hypoperfusion is very intense and meets the criteria of HRS. In the remaining patients, mild or moderate reductions in renal perfusion exist (Fig. 68.1). These latter patients show slightly increased serum creatinine and/or blood urea nitrogen (BUN) levels in baseline conditions (in the absence of diuretic therapy). This moderate renal vasoconstriction is clinically relevant for several reasons: first, it is often associated with marked sodium and water retention and the presence of refractory ascites122; second, it predisposes to the development of HRS23,120,123; and third, it is associated with an impaired survival.62,73
with rapid increase in serum levels of urea and creatinine and low urine volume in some cases, but not all of them. Type 1 HRS is characterized by a rapid and progressive impairment of renal function as defined by a doubling of the initial serum creatinine to a level higher than 2.5 mg per dL in less than 2 weeks. Despite an important reduction of GFR in these patients, serum creatinine levels are commonly lower than values observed in patients with acute renal failure of similar intensity with respect to the reduction in GFR, but without liver disease.125,132,147,148 This is probably due to the lower endogenous production of creatinine secondary to reduced muscle mass in patients with cirrhosis compared with patients without liver disease. Type 1 HRS is associated with a very low survival expectancy, the median survival time being only 2 weeks (Fig. 68.6).146 Type 2 HRS is characterized by a more subtle course with serum creatinine levels around 1.5 to 2.0 mg per dL. Patients are usually in a better clinical condition than those with type 1 HRS and their survival expectancy is longer, approximately 6 months (Fig. 68.6).146 The dominant clinical feature of these patients is diuretic-resistant ascites due to the combination of intense sodium retention, reduced GFR, and marked activation of antinatriuretic systems.122,124,146 Severe spontaneous hyperkalemia is an uncommon feature of HRS. However, marked hyperkalemia may occur if patients are treated with aldosterone antagonists, especially patients with type 1 HRS. Severe metabolic acidosis and pulmonary edema, which are frequent complications of acute renal failure of patients without liver disease, are uncommon findings in patients with HRS. Because HRS is a form of functional renal failure, the characteristics of urine are those of prerenal azotemia, with low urine sodium concentration, and increased urine osmolality and urine-to-plasma osmolality ratio.122,149 Urine volume is not extremely reduced—in a recent series the average urine volume in 60 patients with HRS was 733 mL per day145 and in some cases urine sodium concentration is not extremely reduced.149,150 Table 68.2 shows the current diagnostic criteria of HRS.124
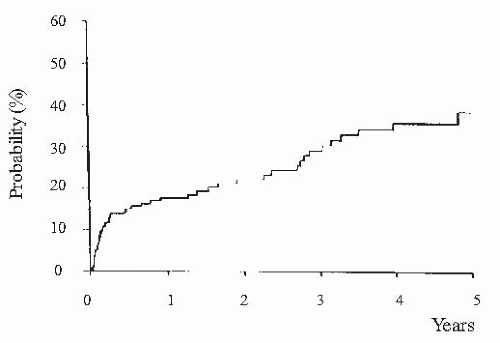 FIGURE 68.5 Probability of developing hepatorenal syndrome in a series of 234 nonazotemic cirrhotic patients with ascites. (Reproduced with permission from Ginès A, Escorsell A, Ginès P, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology. 1993;105:229.) |
TABLE 68.1 Clinical Types of Hepatorenal Syndrome | ||||
|---|---|---|---|---|
|
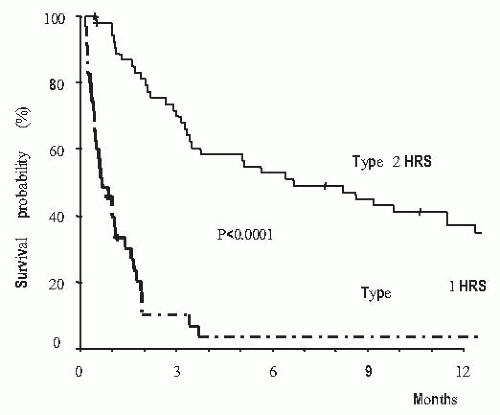 FIGURE 68.6 Survival of patients with cirrhosis according to the type of hepatorenal syndrome. (Reproduced with permission from Alessandria C, Ozdogan O, Guevara M, et al. MELD score and clinical type predict prognosis in hepatorenal syndrome: Relevance to liver transplantation. Hepatology. 2005;41:1282-1289.) |
In addition, several studies have shown that cardiac output is low in patients with HRS, either in absolute values or relative to the reduction in total systemic vascular resistence.151,152,153 This reduction in cardiac output may contribute to the reduction in the effective arterial blood volume and subsequent renal vasoconstriction.152,153,154 In a longitudinal study in patients with cirrhosis it was shown that a reduction in cardiac output was associated with the occurrence of HRS.155 Similarly, in a small series of patients with cirrhosis, those with a low cardiac output had a greater risk of HRS development.156
TABLE 68.2 Diagnostic Criteria of Hepatorenal Syndrome | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
before the diagnosis of HRS is made. Studies in patients with cirrhosis and ascites indicate that drugs that inhibit selectively the cyclooxygenase 2 enzyme (COX-2) do not cause renal failure, at least when administered for a short period of time.164,165 However, studies with longer treatment duration should be performed before these drugs can be confirmed as safe for patients with cirrhosis and ascites. Patients with cirrhosis are also at high risk of developing renal failure due to ATN when treated with aminoglycosides.162,166,167 Because of this high risk of nephrotoxicity and the existence of other effective antibiotics (e.g., third-generation cephalosporins) treatment with aminoglycosides should be avoided in patients with chronic liver disease. Finally, patients with cirrhosis due to hepatitis B and C may also develop renal failure due to glomerulonephritis.168,169,170 In these cases, proteinuria and/or hematuria are almost constant and provide a clue for the diagnosis, which may be confirmed by renal biopsy in selected cases.171
proposed: (1) alterations in the splanchnic and systemic circulation which result in activation of vasoconstrictor and antinatriuretic systems and subsequent renal sodium and water retention; (2) hepatorenal reflex due to increased hepatic pressure which would cause sodium and water retention; and (3) putative antinatriuretic substances escaping from the splanchnic area through portosystemic collaterals that would have a sodium-retaining effect in the kidney. There is a large body of evidence supporting the first of these three pathogenic mechanisms.
TABLE 68.3 Hemodynamic Profile of Patients with Cirrhosis in Different Stages of Disease | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
been the most extensively studied. Increased production of NO, carbon monoxide, glucagon, endocannabinoids, prostaglandins, vasoactive intestinal peptide, adenosine, bile salts, platelet activating factor, substance P, calcitonin gene-related peptide, natriuretic peptides, and adrenomedullin have been proposed as possible factors of the development of splanchnic arterial vasodilation.185,191,205,216,221,222,223,224,225,226,227,228,229,230,231 At present, most available data, obtained mainly from experimental cirrhosis, indicate that NO is the main mediator of arterial vasodilation in cirrhosis (Table 68.4).232 NO synthesis from cirrhotic arterial vessels is markedly increased compared to that of normal vascular tissue. This increased NO synthesis appears to be generalized, except for the intrahepatic circulation, but predominates in the splanchnic territory. Among the different isoforms of NO synthase, the constitutive form appears to be the one responsible for the increased NO synthesis. The normalization of NO synthesis in experimental cirrhosis by the administration of inhibitors of NO synthesis is associated with a marked improvement of splanchnic and systemic hemodynamics, suppression of the increased activity of the RAAS and AVP concentration, increased sodium and water excretion, and reduction or disappearance of ascites.233 So far, only few studies have investigated the effect of acute NO synthesis inhibition in patients with cirrhosis on systemic hemodynamia and/or renal function, with discrepant findings.234,235
Related posts:
 Computed Tomography and Magnetic Resonance Imaging
Computed Tomography and Magnetic Resonance Imaging
 Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
Alport Syndrome, Fabry Disease, and Nail-Patella Syndrome
 Pathophysiology of Nephrotoxic Cell Injury
Pathophysiology of Nephrotoxic Cell Injury
 Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
Immunoglobulin A Nephropathy and Henoch-Schönlein Purpura
 Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
Monoclonal Gammopathies: Multiple Myeloma, Amyloidosis, and Related Disorders
 Peritoneal Dialysis
Peritoneal Dialysis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


