Richard J. Johnson, Jürgen Floege, John Feehally Glomerular disease may have a wide variety of etiologies and clinical presentations (see Chapter 15). Some glomerular diseases are given the generic title of glomerulonephritis (GN), which implies an immune or inflammatory pathogenesis. Although a specific diagnosis can be made in some patients based on clinical presentation and laboratory tests, in most patients a renal biopsy is useful for both classification and prognosis. Ideally, the renal biopsy should be examined by light microscopy, immunofluorescence, and electron microscopy. Using this approach, a histologic pattern can be diagnosed. Some histologic patterns can be coupled with other laboratory tests to identify a specific etiology, but in many cases the condition is idiopathic. However, because treatments are often developed for specific histologic patterns, this approach is currently favored in the management of patients with glomerular disorders. The full assessment of a renal biopsy requires light microscopy, electron microscopy, and examination for deposits of complement and immunoglobulin by immunofluorescence (IF) or immunoperoxidase (IP) techniques. In GN the dominant, but not the only, histologic lesions are in glomeruli (Fig. 16-1). GN is described as focal (only some glomeruli are involved) or diffuse. In any individual glomerulus, injury may be segmental (affecting only part of any glomerulus) or global. Sampling error is possible in a renal biopsy; the extent of a focal lesion may be misjudged in a small biopsy specimen, and sections through glomeruli may miss segmental lesions. Lesions may also be hypercellular because of an increase in endogenous endothelial or mesangial cells (termed proliferative) and an infiltration of inflammatory leukocytes (termed exudative). Severe acute inflammation may produce glomerular necrosis, which is often segmental. The walls of the glomerular capillaries can also be thickened by a number of processes, which include an increase in glomerular basement membrane (GBM) material and immune deposits. Segmental sclerosis and scarring may also occur and are characterized by segmental capillary collapse with the accumulation of hyaline material and mesangial matrix and often with attachment of the capillary wall with Bowman capsule (synechiae or adhesion formation). The classic stains used in light microscopy include hematoxylin-eosin (HE) and the periodic acid–Schiff (PAS) reaction, which is particularly effective for evaluating cellularity and matrix expansion. More specific stains include methenamine silver, which stains GBM and other matrix black and may reveal a double contour to the GBM because of the interposition of cellular material or may show increased mesangial matrix not easily seen with other techniques. Trichrome staining is also useful to show areas of scarring (blue) and immune deposits (red). Crescents are inflammatory collections of cells in Bowman space. Crescents develop when severe glomerular injury results in local rupture of the capillary wall or Bowman capsule, allowing plasma proteins and inflammatory material to enter into the Bowman space. Crescents consist of proliferating parietal and visceral epithelial cells, infiltrating fibroblasts, and lymphocytes and monocytes/macrophages, often with local fibrin deposition. They are called “crescents” because of their appearance when the glomerulus is cut in one plane for histology. These cell collections are destructive and rapidly increasing in size and may lead to glomerular tuft occlusion (Fig. 16-1). If the acute injury is stopped, the crescents may either resolve with restitution of normal morphology or heal by fibrosis, causing irreversible loss of renal function. Crescents are most frequently observed with vasculitis, in Goodpasture disease, and in severe acute GN of any etiology. Tubulointerstitial injury and fibrosis can also accompany GN and may play an important role in the prognosis (see Chapter 79). Indirect IF and IP staining are both used to identify immune reactants (Fig. 16-2). Examination consists of staining for immunoglobulins (IgG, IgA, and IgM), for components of the complement system (usually C3, C4, and C1q), and for the presence of fibrin, which is typically observed in crescents and in capillaries in thrombotic disorders such as hemolytic uremic syndrome and the antiphospholipid syndrome. Immune deposits may occur along the capillary loops or in the mesangium. They may be continuous (linear) or discontinuous (granular) along the capillary wall or in the mesangium. Electron microscopy (EM) is valuable for defining the morphology of the basement membranes, which is abnormal in some forms of hereditary nephropathy (e.g., Alport syndrome and thin basement membrane nephropathy; see Chapter 48), and for identifying fibrils (e.g., in amyloidosis) or tubuloreticular intracellular structures (e.g., in lupus nephritis). EM is also useful for localizing the site of immune deposits, which are usually homogeneous and electron dense (Fig. 16-3). Electron-dense deposits are seen in the mesangium or along the capillary wall on the subepithelial or subendothelial side of the GBM. Infrequently, the electron-dense material lies linearly within the GBM. The sites of immune deposits are helpful in the classification of the types of GN. Proteinuria is the hallmark of glomerular disease. The endothelial glycocalyx and the GBM may repel proteins in part through their highly negative charge (proteins are mostly negatively charged as well) and prevent them from entering Bowman space. A central barrier for protein is the slit diaphragm between the podocyte foot processes1 (Fig. 16-4). The slit diaphragm consists of several transmembrane proteins that extend from adjacent interdigitating foot processes to form a zipper-like scaffold on the outer side of the GBM (Fig. 16-5). The importance of the slit diaphragm in proteinuric states has been documented in numerous hereditary types of nephrotic syndrome in which the mutations involve various slit diaphragm proteins (see Chapter 19). These diseases usually present as a type of steroid-resistant minimal change disease or focal segmental glomerulosclerosis (FSGS). Whereas most recessive mutations of slit diaphragm or podocyte proteins manifest in childhood or even prenatally, dominant mutations tend to manifest in early adult life.2 An exception is steroid-resistant autosomal recessive nephrotic syndrome, in which the homozygous mutation in podocin (NPHS2) presents in childhood, but the heterozygous mutation, when it coexists with the p.R229Q variant polymorphism, may present clinically in young adulthood (age 20 to 40 years old).3 Although it may result from injury or mutation of slit diaphragm proteins, proteinuria may be caused by nonspecific injury to the podocyte in many cases. When the podocyte is injured, it may undergo shape changes with swelling and loss or fusion of the foot processes. Filtration is reduced at sites where the foot processes fuse (possibly accounting for reduction of filtration coefficient Kf seen in nephrotic syndrome), but there are gaps where the podocytes are detached from the GBM. Massive protein filtration may occur at these sites; structurally, the capillary wall defects are likely to correspond to the large pores noted in functional studies4 (Fig. 16-6). Podocyte immaturity can also result in nephrotic syndrome, perhaps from incomplete differentiation and slit diaphragm development. Congenital nephrotic syndrome with mesangial sclerosis has recently been linked with mutations in phospholipase C epsilon gene (PLCE1), which is important in podocyte development.5 In addition to podocyte damage, and in particular slit diaphragm defects, evidence also shows that proteinuria can result from changes in the glomerular endothelium, especially its glycocalyx, as well as from changes in the GBM and altered electrical forces across the GBM. Severe albuminuria reflects a glomerular defect, but some albumin is normally filtered but then endocytosed and metabolized in the proximal tubule or is transcytosed intact through the tubular cell. Proximal tubular dysfunction can therefore result in albuminuria if endocytosis is impaired, although this is generally in the non-nephrotic range.
Introduction to Glomerular Disease
Histologic Classification and Pathogenesis
Histologic Classification
Histopathology
Light Microscopy
Immunofluorescence and Immunoperoxidase Microscopy
Electron Microscopy
General Mechanisms of Glomerular Injury
Proteinuria
Antibody and Antigen
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Introduction to Glomerular Disease
Chapter 16
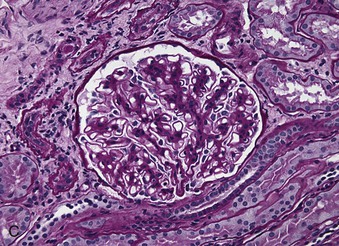
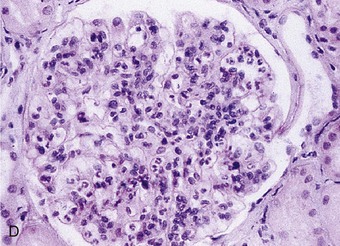
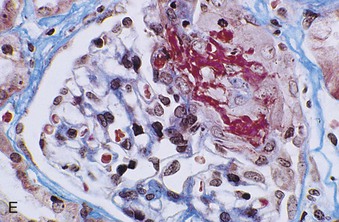
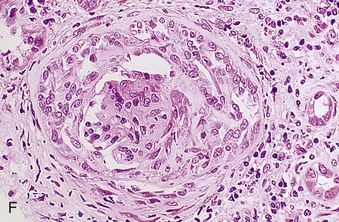
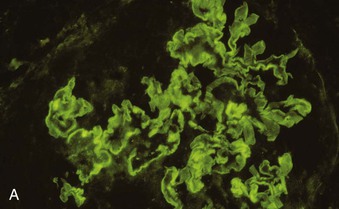
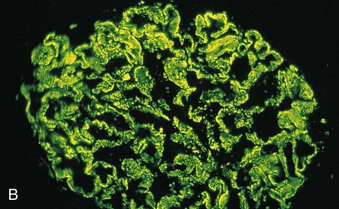
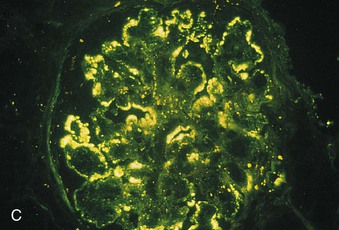
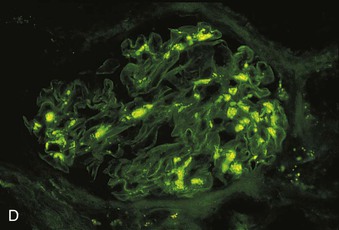
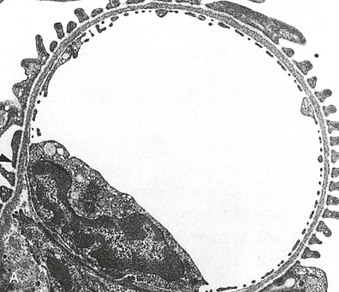
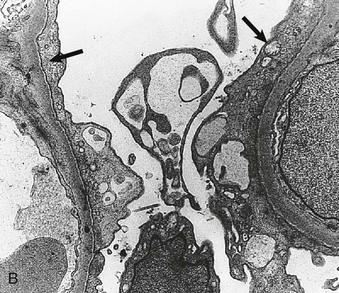
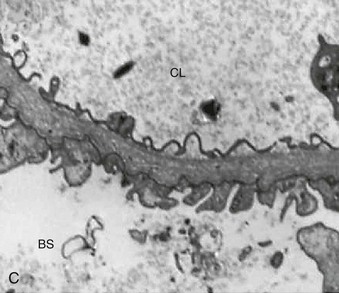
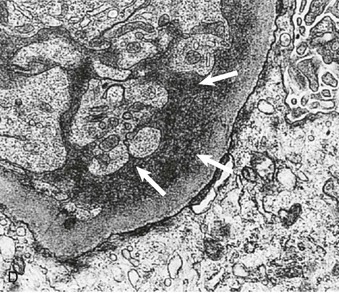
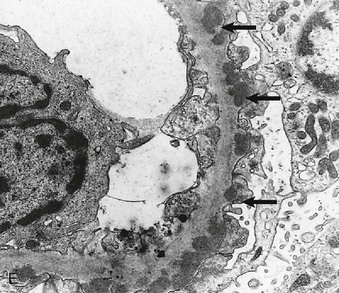
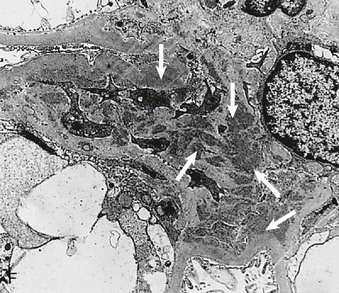
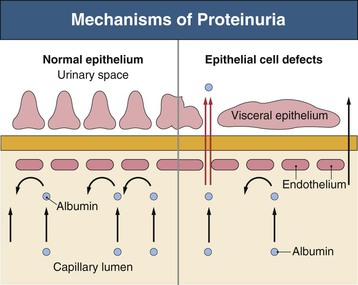
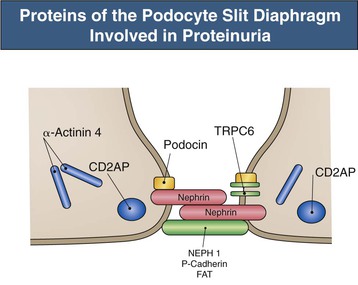
Figure 16-5 Proteins of the podocyte slit diaphragm involved in proteinuria. Several inherited glomerular diseases involve mutations of antigens associated with the slit diaphragm. These include nephrin in congenital nephrotic syndrome of the Finnish type; podocin in autosomal recessive FSGS; and α-actinin and TRPC6 (transient receptor potential cation channel), both associated with autosomal dominant FSGS. In addition, mutation of CD2-associated protein results in nephrotic syndrome in mice. (Modified from reference 1.)
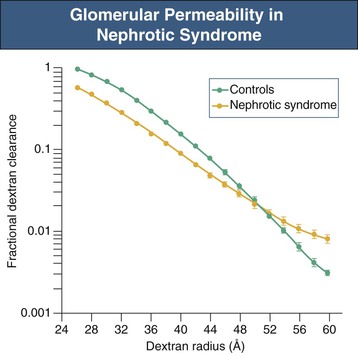
Figure 16-6 Glomerular permeability in nephrotic syndrome. Dextran sieving curve shows the relative glomerular permeability of different-sized dextrans in normal individuals and nephrotic patients with membranous nephropathy and minimal change disease. Nephrotic patients actually have a lower fractional dextran clearance for small dextrans (26 to 48 Å [2.6 to 4.8 nm]) but have an increased clearance for dextrans of larger molecular weight (52 to 60 Å [5.2 to 6.0 nm]). This is consistent with large pores appearing in the GBM. (Modified from reference 4.)






