Generation of Uric Acid
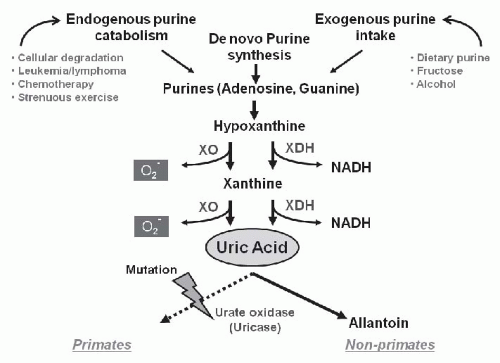
Uric acid is produced from metabolic conversion of either dietary or endogenous purines, primarily in the liver, muscle, and intestine (
Fig. 61.1).
5 Uric acid can also be produced de novo from glycine, glutamine, and other precursors. The immediate precursor of uric acid is xanthine, which is degraded to uric acid by either xanthine oxidase, which generates superoxide anion in the process, or by its isoform, xanthine dehydrogenase, which generates the reduced form of nicotinamide-adenine dinucleotide. Both exogenous purines (such as is present in fatty meat, organ meats, and seafood) and endogenous purines are major sources of uric acid in humans. Approximately two thirds of total body urate is produced endogenously, whereas the remaining one third is accounted for by dietary purines. Purine-rich foods include beer and other alcoholic beverages, anchovies, sardines in oil, fish roes, herring, organ meat (liver, kidneys, sweetbreads), legumes (dried beans, peas), meat extracts, consommé, gravies, mushrooms, spinach, asparagus, and cauliflower.
6 In healthy men, the urate pool averages about 1,200 mg with a mean turnover rate of 700 mg per day.
Excretion of Uric Acid
The primary site of excretion of uric acid is the kidney, with normal urinary urate excretion in the range of 250 to 750 mg per day. Although urate (the form of uric acid at blood pH of 7.4) is freely filtered in the glomerulus, there is evidence that there is both reabsorption and secretion in the proximal tubule, and as a consequence the fractional urate excretion is only 8% to 10% in the normal adult because urate reabsorption dominates oversecretion in the kidney. Some adaptation occurs with renal disease, in which the fractional excretion will increase to the 10% to 20% range. In addition, uric acid is also removed by the gut, where uric acid is degraded by uricolytic bacteria, and this may account for one third of the elimination of uric acid in the setting of renal failure.
The historic paradigm of uric acid excretion consists of a four-step model with glomerular filtration, followed by reabsorption, secretion, and postsecretory reabsorption, the latter three processes all occurring in the proximal convoluted tubule.
7,
8 However, ideas of the handling of uric acid by the kidney have changed greatly during the last decades, with characterization and isolation of transporters and channels mainly or exclusively restricted to urate transport (
Fig. 61.2).
9,
10 Membrane vesicle studies have suggested the existence of two major mechanisms modulating urate reabsorption and secretion, consisting of a voltage-sensitive pathway and a urate/organic anion exchanger. Recently several of these transporters/channels have been identified. Organic anion transporters 1-10 (OAT1-10) and the urate transporter-1 (URAT-1) belong to the SLC22A gene family and accept a huge variety of chemically unrelated endogenous and exogenous organic anions including uric acid. Endou’s group identified URAT-1, which is encoded by SLC22A12, as the major organic anion exchanger for uric acid on the apical (luminal brush border) side of the proximal tubular cell.
9 In the human kidney, urate is transported via URAT-1 across the apical membrane of proximal tubular cells, in exchange for anions being transported back into the tubular lumen to maintain electrical balance. URAT-1 has a high affinity for urate together with lactate, ketones, a-ketoglutarate, and related compounds. Pyrazinamide, probenecid, losartan, and benzbromarone all inhibit urate uptake in exchange for chloride at the luminal side of the cell by competition with the urate exchanger. OAT-4 exhibits 53% amino acid homology with URAT1.
Urate then moves across the basolateral membrane into the blood by other organic anion transporters, of which the most important is SLC2A9 (also known as GLUT9).
11,
12 GLUT-9 is highly expressed in the kidney and liver. GLUT-9L (long isoform) is localized to basolateral membranes in proximal tubule epithelial cells, whereas the splice variant GLUT-9S (short isoform) localizes to apical membranes (
Fig. 61.2).
13 Vitart et al.
14 showed that GLUT-9 transports urate and fructose, using a
Xenopus oocyte expression system. GLUT-9 deficiency resulted in renal hypouricemia and is consistent with GLUT-9 being an efflux transporter of intracellular urate from the tubular cell to the interstitium/blood space.
15 Efflux transport of urate at basolateral membranes appears to depend principally on GLUT-9L whereas URAT-1 mainly acts as an influx transporter for urate at apical membranes.
OAT-4 and OAT-10 function as an organic anion/dicarboxylate exchanger and are responsible for the reabsorption of organic anions driven by an outwardly directed dicarboxylate gradient.
16 In addition, OAT1 and OAT3 may have a role in the transport of urate from the blood into the proximal tubule.
17,
18Urate secretion appears to be mediated principally by a voltage-sensitive urate transporter, which is expressed ubiquitously and localizes to the apical side of the proximal tubule in the kidney. Genomewide association studies revealed the region which is related to serum urate concentration.
19 Recently, a novel human renal apical organic anion efflux transporter, called MRP4, has been identified.
20 MRP4 is a member of the ATP-binding cassette transporter family. It is proposed to mediate secretion of urate and other organic anions such as cAMP, cGMP, and methotrexate across the apical membrane of human renal proximal tubular cells. Human MRP4 is an ATP-dependent unidirectional efflux pump for urate with multiple allosteric substrate binding sites.
21 Renal sodium-dependent phosphate transport protein-1 (NPT-1),
22 which was first cloned as a phosphate transporter, is located in the proximal
convoluted renal tubule (
Fig. 61.2). NPT1 mediates voltage-sensitive transport of organic anions, including urate, and is suggested to function as a urate secretor.
23 Another transporter located at the apical membrane of proximal tubules is ATP-binding cassette, sub-family G, member 2 (ABCG2). The ability of ABCG2 to transport urate was recently confirmed by measuring urate efflux from ABCG2-expressing
Xenopus oocytes.
24Another gene involved in renal transport of urate is Tamm-Horsfall protein (THP), also known as uromodulin. THP is exclusively expressed and secreted by epithelial cells of the thick ascending limb, where it has been shown to have antibacterial effects. THP also co-localizes with the Na-K-2Cl transporter in lipid rafts in the apical cell membrane, suggesting a functional interaction.
25 Mutations in the human uromodulin gene have been identified in subjects with medullary cystic kidney disease type 2 and in patients with familial juvenile hyperuricemic nephropathy (see subsequent text).
26,
27 It is not yet known how the THP mutation leads to hyperuricemia, as most evidence suggests that uric acid handling is restricted to the proximal tubule. However, there is some evidence that some urate secretion in the rat can occur distal to the proximal tubule.
28 Furthermore, there is also some evidence that the THP mutation may lead to sodium and water wasting, possibly resulting in stimulating urate reabsorption proximally (see following section on Familial Juvenile Hyperuricemic Nephropathy).
Causes of Hyper- and Hypouricemia
Hyperuricemia has been arbitrarily defined as >7.0 mg per dL in men and >6.5 mg per dL in women. “Normal” serum uric acid levels in the population appear to be rising throughout the last century, likely as a consequence of changes in diet, and mean levels in men in the United States are now in the 6.0 to 6.5 mg per dL range.
4 Uric acid levels tend to be higher in certain populations (e.g., African American and Pacific Islanders), with certain phenotypes (obesity, metabolic syndrome) and with special diets (meat eaters).
4 Uric acid also has a circadian variation, with the highest levels in the early morning.
29The serum urate concentration reflects the balance between urate production and elimination. Hyperuricemia may occur from excessive production of urate (overproduction) or decreased elimination (underexcretion), and frequently a combination of both processes occur in the same patient. Furthermore, uric acid levels may vary in the same
individual by as much as 1 to 2 mg per dL during the course of a day, due to the effects of diet and exercise.
Genetic mechanisms mediating hyperuricemia include overproduction due to mutations of two enzymes: hypoxanthine-guanine phosphoribosyltransferase (HGPRT) and phosphoribosyl pyrophosphate synthetase (PRPPS) (
Table 61.1). Subjects with Lesch-Nyhan syndrome (due to a mutation of HGPRT on the X chromosome) present in childhood with neurologic manifestations (mental retardation, choreoathetosis, and dystonia) and have an increased risk for nephrolithiasis, renal failure, and gout. A partial deficiency of HGPRT may manifest later in life as recurrent gout and/or nephrolithiasis (partial HGPRT deficiency (Kelley-Seegmiller syndrome).
30 Other genetic mechanisms include subjects with the uromodulin mutation, who develop hyperuricemia (due to underexcretion) with early and progressive renal disease (see subsequent text). Certain populations such as indigenous peoples living in Oceania also have higher uric acid levels than Caucasian populations.
31 Finally, African Americans also have higher uric acid levels and a twofold higher incidence of gout compared to Caucasian or Asian populations
32; however, this could also reflect diets higher in fructose-containing sugars (see subsequent text) rather than genetic mechanisms.
Hyperuricemia may also result from diets high in purines, from ethanol, and from fructose. The effect of alcohol is in part related to increased urate synthesis, which is due to enhanced turnover of ATP during the conversion of acetate to acetyl-CoA as part of the metabolism of ethanol.
33 In addition, acute alcohol consumption causes lactate production, and because lactate is an antiuricosuric agent, it will reduce renal urate excretion and exacerbate hyperuricemia.
34 Fructose (a simple sugar present in sucrose, table sugar, high fructose corn syrup, honey, and fruits) can also induce a rapid rise in serum uric acid, due in part to its rapid phosphorylation in hepatocytes with the stimulation of AMP deaminase and ATP consumption.
35 Chronic fructose consumption also stimulates uric acid synthesis.
35 It has been proposed that the marked increase in fructose intake may have a role in the rising levels of serum uric acid and obesity worldwide.
36Uric acid may also be affected by exercise, with moderate exercise reducing urate levels (probably by increasing renal blood flow) and severe exercise causing a rise in uric acid (probably due to ATP consumption with adenosine and xanthine formation). Urate levels vary among gender, in that premenopausal women have lower uric acid, a fact attributed to the uricosuric effect of estrogen.
37 The mechanism may relate to gender effects on URAT-1 expression, as recent studies suggest that male mice have higher URAT-1 expression in their proximal tubules compared to female mice.
38 Androgens also increase xanthine oxidase levels that might contribute to the higher uric acid levels observed in men.
39 Uric acid also tends to increase in the setting of low blood volume and/or low salt diet (due to increased proximal reabsorption), and following the administration of catecholamines or angiotensin II (due to renal vasoconstriction resulting in increased reabsorption). Urate production also relates to body size and weight, so that larger persons produce more urate than those who are smaller. Hyperuricemia is particularly common in the obesity and metabolic syndrome (thought to be secondary to the effect of insulin to stimulate uric acid reabsorption)
40 and in untreated hypertension (thought to be due to reduced renal blood flow).
41 Thiazides also increase uric acid reabsorption by decreasing blood volume and via direct interaction with the organic anion exchanger.
Other drugs (cyclosporine, pyrazinamide, low dose aspirin) also increase uric acid, primarily by interfering with renal excretion. In addition, the generation of organic anions such as lactate, β-hydroxybutyrate, and others may interfere with urate secretion in the proximal tubule and cause a rise in serum uric acid. Chronic lead ingestion can also cause hyperuricemia by reducing urate excretion, whereas high concentrations tend to cause proximal tubular injury with no rise in uric acid.
Uric acid is also increased in the setting of tissue hypoxia
42 or with cell turnover.
43 With tissue hypoxia, ATP is consumed
and the isoform, xanthine oxidase, is induced, resulting in increased local uric acid concentrations. Uric acid levels are thus high in subjects with congestive heart failure, high altitude hypoxia, congenital cyanotic heart disease, and with obstructive sleep apnea. Uric acid levels are commonly elevated with certain malignancies, especially leukemias and lymphomas, and levels may sharply rise following chemotherapy (see acute urate nephropathy in the following text).
44 Finally, uric acid has a tendency to be elevated in polycythemia vera and other myeloproliferative disorders.
45In the setting of reduced renal function, the fractional excretion of urate increases but is not enough to fully compensate for the reduction in glomerular filtration rate (GFR), and as a consequence serum uric acid levels rise. Conversely, uric acid excretion via the gastrointestinal tract is also enhanced,
46 and therefore serum uric acid levels tend to be only mildly elevated in patients with chronic renal disease, and gout is relatively rare.
Low uric acid levels (levels <2.0 mg per dL) can occur via a variety of mechanisms, including with liver disease (due to decreased production), Fanconi syndrome (due to impaired proximal tubular function), and with diabetic glycosuria (due to proximal tubular dysfunction) (
Table 61.2). Drugs such as probenecid, high-dose salicylates, sulfinpyrazone, benziodarone, benzbromarone, and losartan are all uricosuric, whereas allopurinol, febuxostat, and oxypurinol lower uric acid by blocking xanthine oxidase. Statins also lower uric acid,
47 and recombinant uricase (rasburicase) can markedly reduce serum uric acid and is approved for use in children with tumor lysis syndrome (in which marked hyperuricemia may develop).
48 There is also a hereditary hypouricemia syndrome that has been observed, and is particularly common in Japan, where it has been shown to be due to a mutation in the URAT-1 gene.
49 A similar hypouricemia syndrome has also been observed with mutations in SLC2A9.
50 These patients are particularly prone to develop acute renal failure following vigorous exercise, in which it is postulated to be due to massive uricosuria following ATP consumption in the muscle.