fever is present in one-quarter of patients at the diagnosis, but can often be seen as a consequence of plasma exchange. Less common manifestations include acute abdomen, pancreatitis, and sudden death.11
TABLE 55.1 Classification of Hemolytic Uremic Syndrome (HUS) and Thrombotic Thrombocytopenic Purpura (TTP) According to Clinical Presentation and Underlying Etiology | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
erythrocytes is crucial to confirm the microangiopathic nature of the hemolytic anemia, provided that heart valvular disease and other anatomic artery abnormalities that may cause erythrocyte fragmentation are excluded. Other indicators of intravascular hemolysis include elevated lactate dehydrogenase (LDH), increased indirect bilirubin, and low haptoglobin levels.5,11,12 The Coombs test is negative. Moderate leukocytosis may accompany the hemolytic anemia. Thrombocytopenia is uniformly present in HUS and TTP. It may be severe, but is usually less so in patients with predominant renal involvement.13 The presence of giant platelets in the peripheral smear or reduced platelet survival time (or both) is consistent with peripheral consumption. In children with Stx-HUS, the duration of thrombocytopenia is variable and does not correlate with the course of renal disease.14 Bone marrow biopsy specimens usually show erythroid hyperplasia and an increased number of megakaryocytes. Prothrombin time (PT), partial thromboplastin time (PTT), the fibrinogen level, and coagulation factors are normal, thus differentiating HUS and TTP from disseminated intravascular coagulation (DIC). Mild fibrinolysis with minimal elevation in fibrin degradation products, however, may be observed.
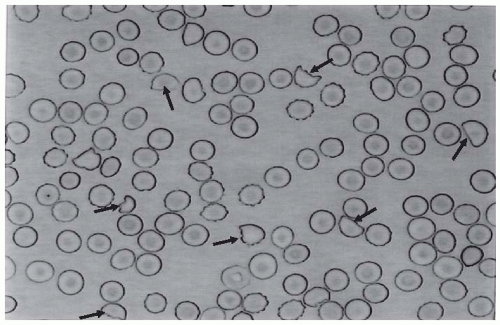 FIGURE 55.1 A peripheral blood smear from a patient with thrombotic microangiopathy. The presence of fragmented red blood cells that may acquire the appearance of a helmet (fragmented erythrocytes with the shape of a helmet are identified by the black arrows) is pathognomonic for microangiopathic hemolysis in patients with no evidence of heart valvular disease. |
of swelling and detachment of the endothelial cells from the basement membrane and the accumulation of fluffy material in the subendothelium (Figs. 55.2 and 55.3), intraluminal platelet thrombi, and a partial or complete obstruction of the vessel lumina (Fig. 55.4).16,17,18 These lesions are similar to those seen in other renal diseases such as scleroderma, malignant nephrosclerosis, chronic transplant rejection, and calcineurin inhibitor nephrotoxicity. In HUS, microthrombi are present primarily in the kidneys, whereas in TTP they mainly involve the brain, where thrombi may repeatedly form and resolve, producing intermittent neurologic defi-cits. In pediatric patients, particularly in those younger than 2 years of age, and in those with HUS secondary to gastrointestinal infection with Stx-producing strains of E. coli , the glomerular injury is predominant.16
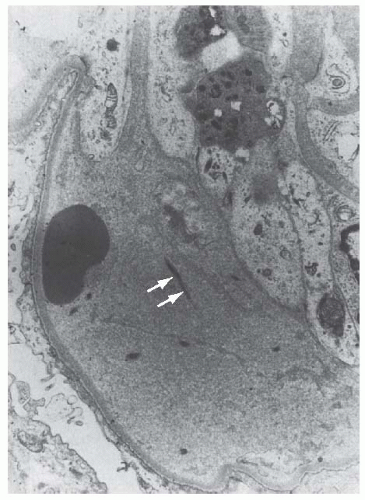 FIGURE 55.2 The detachment of an endothelial cell from the underlying glomerular basement membrane in a case of hemolytic-uremic syndrome. A red blood cell is in close contact with the glomerular basement membrane. Electron-lucent “fluffy” material and a few strands of fibrin (arrows) are present in the subendothelial space (×7,000). (Courtesy of Drs. C. L. Pirani and V. D’Agati.) |
and tissues (Fig. 55.8). These thrombi consist of fibrin and platelets, and their distribution is widespread. They are most commonly detected in the kidneys, the pancreas, the heart, the adrenals, and the brain. Compared to HUS, pathologic changes of TTP are more extensively distributed, probably reflecting the more systemic nature of the disease.16,17,18
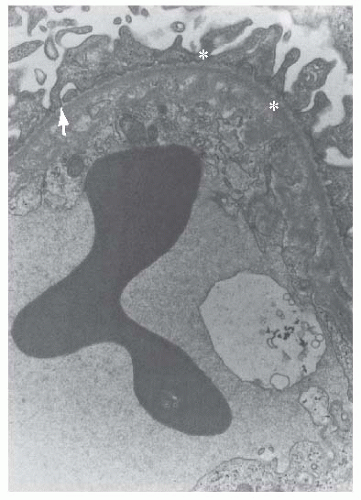 FIGURE 55.3 Electron-lucent “fluffy” material (arrow) with some electron-dense deposits (asterisks) are located between the cytoplasm of an endothelial cell and the glomerular basement membrane in a segment of glomerular capillary from a patient with hemolytic-uremic syndrome (×12,000). (Courtesy of Drs. C. L. Pirani and V. D’Agati.) |
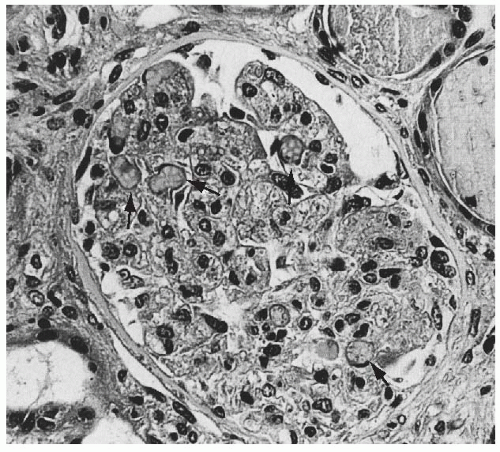 FIGURE 55.4 Swelling of the glomerular endothelial cells and occlusion of almost all capillary lumens packed with red blood cells (arrows) in a case of hemolytic-uremic syndrome (Trichrome, × 250). |
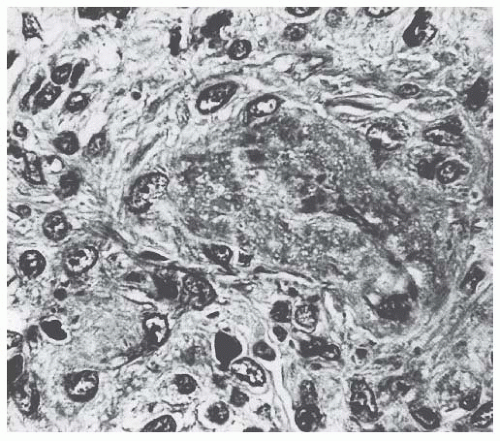 FIGURE 55.5 Thrombotic and necrotic changes in a small artery from an adult patient with hemolytic-uremic syndrome. (Trichrome, ×375.) |
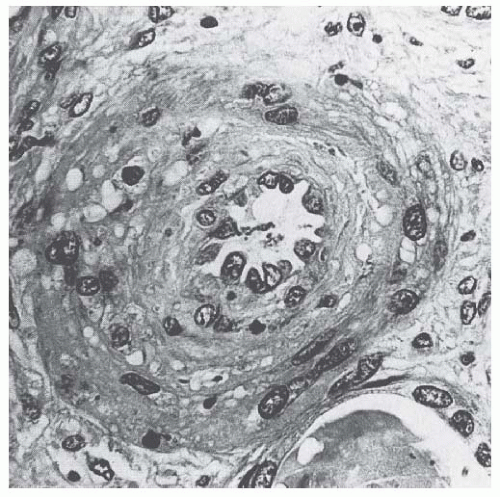 FIGURE 55.6 Occlusion of an interlobular artery with intimal swelling and myointimal proliferation in a case of adult hemolytic-uremic syndrome. (Trichrome,× 375.) |
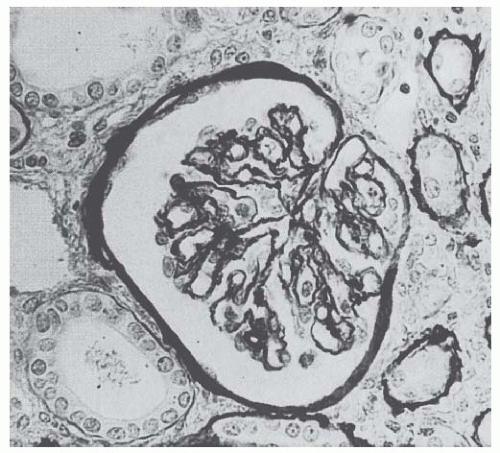 FIGURE 55.7 Ischemic glomerular lesions characterized by thickening and wrinkling of glomerular capillary walls and atrophy of the glomerular tuft in a case of adult hemolytic-uremic syndrome. (Silver, ×250.) |
such as O111:H8, O103:H2, O123, O26, and the O104:H4 strain isolated in the recent German outbreak)20,21 isolated from human cases with diarrhea were found to produce a toxin similar to the one of S. dysenteriae . After food contaminated by Stx-producing E. coli or S. dysenteriae is ingested, the toxin is released into the gut and may cause watery or, most often, bloody diarrhea because of a direct effect on the intestinal mucosa. Stx-producing E. coli closely adhere to the epithelial cells of the gastrointestinal mucosa causing the destruction of the brush border villi.22 Stxs are picked up by polarized gastrointestinal cells via transcellular pathways and translocate into the circulation,23 probably facilitated by the transmigration of neutrophils,24 which increase paracellular permeability. Circulating human blood cells, such as erythrocytes,25 platelets,26,27 and monocytes,28 express Stx receptors on their surface and have been suggested to serve as Stx carriers from the intestine to the kidney and other target organs.
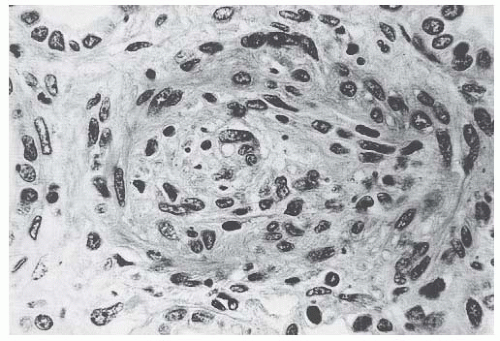 FIGURE 55.8 Marked endothelial and myointimal cell proliferation with occlusion of the lumen of an interlobular artery in a case of thrombotic thrombocytopenic purpura (Trichrome, ×375.) |
were protected against glomerular abnormalities and renal function impairment, indicating the involvement of complement activation, via the alternative pathway, in the glomerular thrombotic process in HUS mice.46
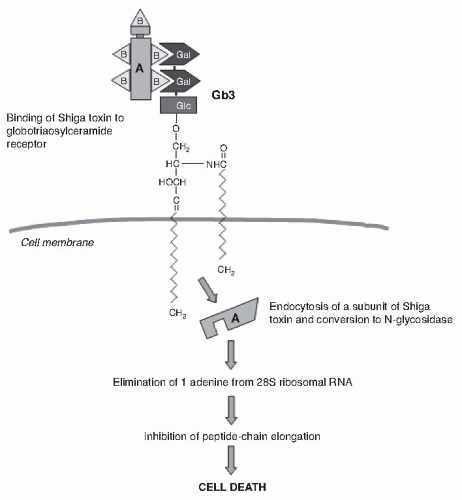 FIGURE 55.9 The binding and mechanism of action of Shigalike toxin. The B subunits of Shiga toxin molecules attach to galactose (Gal) disaccharides of globotriaosylceramide (Gb3) receptors on the membrane of monocytes, polymorphonuclear cells, platelets, glomerular endothelial cells, and tubular epithelial cells. The toxin is internalized via retrograde transport through the Golgi complex. Then the A and B subunits dissociate, and the A subunit is translocated to the cytosol. The A subunit blocks peptide chain elongation by eliminating one adenine from the 28S ribosomal RNA. |
complicated by HUS is self-limiting and is not associated with an increased long-term risk of high blood pressure or renal dysfunction, as shown by a 4-year follow-up study in 951 children who were exposed to a drinking water outbreak of E. coli O157:H7.50
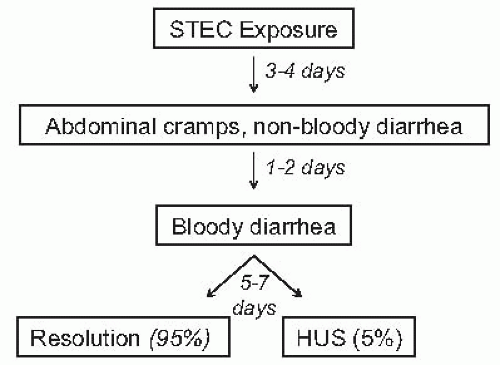 FIGURE 55.10 Timing of the events that may follow exposure to Shiga toxin-producing E. coli. HUS, hemolytic uremic syndrome. |
E. coli O157:H7 a selective advantage if these organisms are not as readily eliminated from the bowel as are the normal intestinal flora. This might specifically apply to the O104:H4 strain, which has acquired a host of new resistances to antibiotics most commonly used in human diseases, such as cephalosporins, monobactams, fluoroquinolones, cotrimoxazole, tetracyclines, and aminoglycosides, which are in large part mediated by ESBL.20,63 Actually, ESBL-mediated resistances might offer to this strain a selective advantage over the normal intestinal flora upon exposure to one or more of the previous antimicrobials administered at the onset of gastrointestinal symptoms.57 Moreover, several antimicrobial drugs, particularly the quinolones, trimethoprim, and furazolidone, are potent inducers of the expression of the Stx2 gene and may increase the level of toxin in the intestine. Although the possibility of a cause-and-effect relationship between antibiotic therapy and an increased risk of HUS has been challenged by a recent meta-analysis of 26 reports,64 there is no reason to prescribe antibiotics because they do not improve the outcome of colitis, and bacteremia is only exceptionally found in Stx- associated HUS. However, when hemorrhagic colitis is caused by Shigella dysentery type 1, early and empirical antibiotic treatment shortens the duration of diarrhea, decreases the incidence of complications, and reduces the risk of transmission by shortening the duration of bacterial shedding. Thus, in developing countries where Shigella is the most frequent cause of hemorrhagic colitis, antibiotic therapy should be started early and even before the involved pathogen is identified. Whether early treatment with carbapenems or antimicrobials, such as fosfomycin, which are electively effective against ESBL-producing bacteria,65 may help prevent progression from enterocolitis to HUS in patients with evidence or suspicion of gastrointestinal infection with E. coli O104:H4 or other ESBL-producing strains may merit formal investigation.
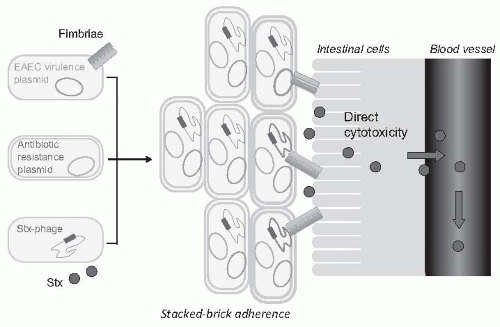 FIGURE 55.11 The hypothetical origin of the O104:H4 E. coli strain isolated from the German outbreak. An ancestral enteroaggregative E. coli (EAEC) strain with plasmids encoding for different virulence factors, including fimbriae that help stick to the intestinal cell, might have acquired the Shiga toxin phage (Stx-phage) characteristic of enterohemorragic E. coli (EHEC) and plasmids encoding for expanded- spectrum betalactamases. Close adhesion to the intestinal cell would facilitate the uptake of the Shiga toxin into the bloodstream, whereas resistance to most commonly used antibiotics would offer selective advantage over the normal intestinal flora upon antibiotic exposure. |
lead us to consider plasma infusion or exchange suitable for adult patients, in particular in those with severe renal insuf- ficiency and central nervous system involvement.
TABLE 55.2 Specific Therapies Used in Thrombotic Microangiopathy, Dosing, and Efficacy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
in 5% to 10% of renal transplant patients who receive cyclosporine and in approximately 1% of those who are given tacrolimus.87,88,89 TMA usually develops in the first weeks posttransplant when patients are treated with high doses of the immunosuppressant. Plasma exchange, combined with dose reduction or the withdrawal of calcineurin inhibitors, achieved remission in up to 80% of patients with de novo posttransplant HUS.88
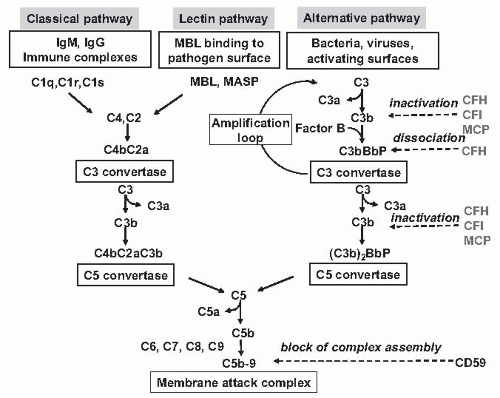 FIGURE 55.12 The Three activation Pathways fo Complement. The classical pathway is initiated by the binding of the C1 complex to antibodies bound to an antigen on the surface of a bacterial cell, leading to the formation of a C4b2a enzyme complex, the C3 convertase of the classical pathway. The mannose-binding lectin pathway is initiated by binding of the complex of mannose-binding lectin (MBL) and the serine proteases mannose-binding lectin-associated proteases 1 and 2 (MASP1 and MASP2) to mannose residues on the surface of a bacterial cell, which leads to the formation of the C3 convertase enzyme C4bC2a. The alternative pathway is initiated by the covalent binding of a small amount of C3b generated by spontaneous hydrolysis in plasma to hydroxyl groups on cell-surface carbohydrates and proteins. This C3b binds factor B to form the alternative pathway C3 complex C3bBb. The C3 convertase enzymes cleave many molecules of C3 to form the anaphylatoxin C3a and C3b, which binds covalently around the site of complement activation. Some of this C3b binds to C4b and C3b in the convertase enzymes of the classical and alternative pathways, respectively, forming C5 convertase enzymes that cleave C5 to form the anaphylatoxin C5a and C5b, which initiates the formation of the membrane-attack complex. The human complement system is highly regulated as to prevent nonspecific damage to host cells and to limit the deposition of complement to the surface of pathogens. This fine regulation occurs through a number of membraneanchored and fluid phase regulators that inactivate complement products formed at various levels in the cascade and that protect host tissues. (See Color Plate.) CFB, complement factor B; CFI, complement factor I; CFH, complement factor H; MCP, membrane cofactor protein; CD59, protectin (prevents the terminal polymerization of the membrane attack complex). |
that protect against invading organisms.94 Three activation pathways—classical, lectin, and alternative pathways— produce protease complexes, termed C3 and C5 convertases that cleave C3 and C5, respectively, eventually leading to the membrane attack complex (MAC or C5b-9) that causes cell lysis (Fig. 55.12). The alternative pathway is initiated spontaneously in plasma by C3 hydrolysis, which is responsible for covalent deposition of a low amount of C3b onto practically all plasma-exposed surfaces. On the bacterial surface, C3b leads to opsonization for phagocytosis by neutrophils and macrophages. Without regulation, a small initiating stimulus is quickly amplified to a self-harming response until the consumption of complement components. On host cells, such a dangerous cascade is controlled by membrane-anchored and fluid-phase regulators (Fig. 55.12). They both favor the cleavage of C3b to inactive iC3b by the plasma serine-protease factor I (CFI, cofactor activity) and dissociate the multicomponent C3 and C5 convertases (decay acceleration activity). Foreign targets and injured cells that either lack membrane-bound regulators or cannot bind soluble regulators are attacked by complement.
TABLE 55.3 Outcome of Atypical Hemolytic Uremic Syndrome According to the Associated Genetic Abnormality
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|





